Appearance
体内重编程过早终止可通过改变表观遗传调控导致癌症发生(2014)
论文信息
教授: Yasihiro Yamada · 大学: 东京大学 · 阅读日期: 2026-03-10 ~ 2026-03-17
基本信息
| 项目 | 内容 |
|---|---|
| 标题 | Premature Termination of Reprogramming In Vivo Leads to Cancer Development through Altered Epigenetic Regulation |
| 作者 | Kotaro Ohnishi et al. |
| 发表于 | Cell |
| DOI/链接 | 原文 |
| 被引次数 | - |
总结概要
在小鼠内,使用Dox(多西环素)控制重编程状态,结果发现重编程过程被打乱的细胞变成了癌细胞——而DNA序列完全没有变化。证明了:光是表观遗传出问题,不需要DNA损坏,就能得癌症。
研究背景与动机
- 传统观点认为:癌症 = 基因突变的逐步累积(即Vogelstein的多步致癌模型),但是作者发现iPS细胞的产生过程(重编程)不需要改变DNA序列,却能让体细胞获得无限增殖能力——这恰恰是癌细胞的核心特征
- 如果重编程过程在中途失败(没有完全变成iPS细胞),那些"半成品"细胞会不会变成癌细胞?如果是的话,这是否意味着不需要基因突变,仅靠表观遗传改变就能致癌?
核心方法
整体思路
制造模型——观察结果——证实
- 即做出Reprogrammable小鼠
- 组织学+免疫染色(H&E染色、Ki67、免疫荧光);
- 基因/蛋白表达检测(qRT-PCR、微阵列芯片)
- 表观遗传/基因组分析(RRBS甲基化测序、CGH)
制作模型(Tet-on系统)
目的:造一只小鼠,让它体内的细胞可以被药物(Dox)遥控,随时开启或关闭重编程因子的表达。
中心法则:
正常情况下rtTA(激活蛋白)无法启动TepOP(四环素应答元件),从而无法表达OSKM重编程因子,然而在Dox多西环素的作用下,Dox与rtTA结合使其能够启动TepOP,表达OSKM重编程因子
无Dox: rtTA(软)--X--→ TetOP → OSKM 不表达
有Dox: Dox + rtTA(硬)---→ TetOP → OSKM 表达!
撤Dox: rtTA(软)--X--→ TetOP → OSKM 停止表达路人小鼠的养成方法(如何将rtTA + TetOP-OSKM这一系统装进小鼠)
a. KH2 ESC是一种特殊的"预制"干细胞,已经在基因组的两个安全位置预留好了"插槽": 插槽1(Rosa26位点): 已经装好了rtTA,永远表达,全身各组织都有 插槽2(Col1a1位点): 空的着陆点,等着把TetOP-OSKM装进去
操作过程:
- 把含有TetOP-OSKM-mCherry的DNA载体(一个环形DNA分子)和FLP重组酶质粒,混在一起
- 用**电穿孔(electroporation)**送入ESC——简单理解就是用短暂的电脉冲在细胞膜上打小孔,让外面的DNA分子进入细胞内
- FLP重组酶(一种"分子剪刀")识别载体上和基因组Col1a1位点上的特定标记序列(FRT),像拉拉链一样把载体DNA精确嵌入到Col1a1位点
- 用G418药物筛选——只有成功整合的细胞能活下来(因为整合同时恢复了一个抗药基因)
用ESC造小鼠,方法叫囊胚注射(blastocyst injection):
- 从普通怀孕母鼠体内取出囊胚(受精后约3.5天的早期胚胎,此时是一个中空的球,约由几十个细胞组成)
- 用极细的玻璃针,把10-15个装好转基因的ESC注射到囊胚的内部
- 把注射后的囊胚移植回另一只假孕母鼠的子宫
- 等待出生
实验流程
普通饮水 → 换成含Dox的水(开启重编程)→ 过N天 → 换回普通水(关闭重编程)→ 观察结果论文中不同的实验方案就是改变N的天数:
- N = 28天(持续开启)→ 形成畸胎瘤(完全重编程成功)
- N = 3-5天 → 出现异型增生,但撤药后恢复正常
- N = 7天 → 撤药后,肿瘤继续生长!(这就是核心发现)
实验与结果
观察形态
方法1:H&E染色
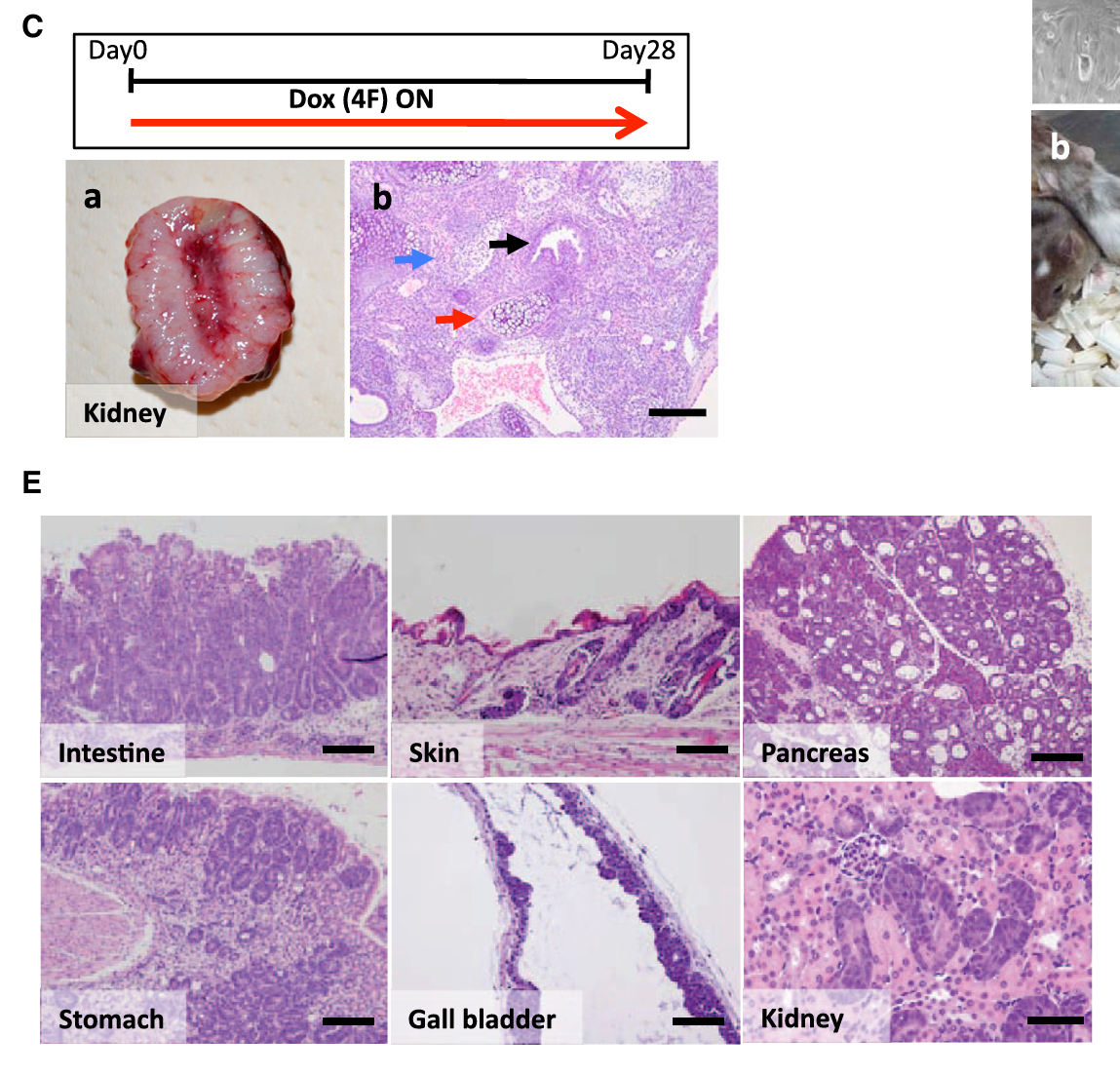
- Fig 1C:Dox给药28天后肾脏肿瘤的切片,看到三个胚层分化(神经组织、软骨、腺上皮)→ 病理诊断:畸胎瘤
- Fig 1E:Dox给药3-9天,肠、皮肤、胰腺、胃、胆囊、肾脏都出现了异型增生——细胞排列紊乱、核浆比增大、极性消失
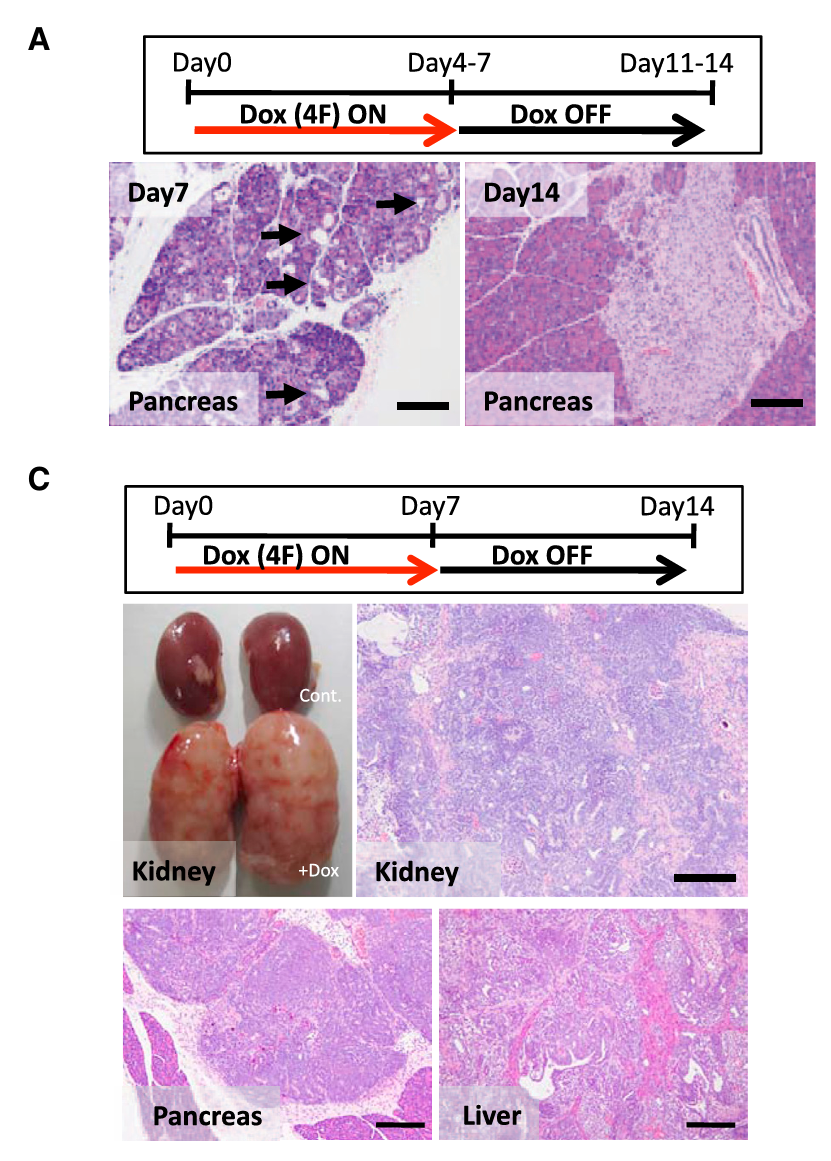
- Fig 2A:Dox 7天→撤除7天,胰腺恢复正常组织形态
- Fig 2C:Dox 7天→撤除7天,肾脏出现侵袭性肿瘤
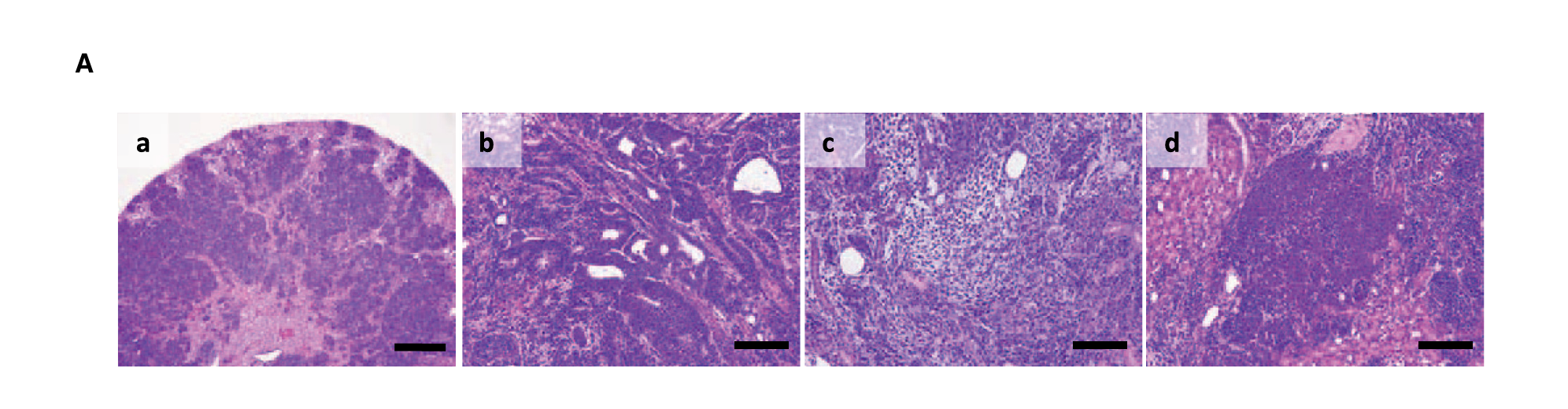
- Fig 5A:肾脏肿瘤的详细病理——包含上皮成分、间质成分、胚芽样成分,这三种成分共存是Wilms瘤的经典病理特征
方法2:免疫组织化学/免疫荧光
❤这一部分可以链接笔记免疫荧光&免疫组化部分的内容
Fig 1F(Ki67)、1G(Oct3/4、2A)、2B(Insulin、BrdU)、3E-F(GFP、Lin28b、Oct3/4)
原理:
组织切片上的目标蛋白(抗原)
↓ 加入针对该蛋白的特异性抗体(一抗)
一抗结合到目标蛋白上
↓ 加入带有"标记"的二抗(二抗能识别一抗)
二抗结合到一抗上,把"标记"带到了目标蛋白的位置
↓ 显色或发光
你就能在显微镜下看到目标蛋白在哪些细胞里免疫组织化学(IHC)= 用酶显色
- 二抗连接的是一种酶(通常是辣根过氧化物酶HRP)
- 加入底物(DAB)→ 酶催化底物变成棕色沉淀
- 在普通光学显微镜下观察
- 优点:可以同时看到组织结构(因为背景是H&E的蓝/粉色)
- 论文中Fig 1F的Ki67就是这种——棕色的细胞核 = Ki67阳性 = 正在增殖
免疫荧光(IF)= 用荧光标记
- 二抗连接的是荧光分子(如绿色的FITC、红色的Cy3、蓝色的DAPI标记细胞核)
- 在荧光显微镜/共聚焦显微镜下观察
- 优点:可以同时看多种蛋白(不同蛋白用不同颜色的荧光标记)
- 论文中Fig 1G就是这种——绿色=Oct3/4,红色=2A肽,蓝色=细胞核(DAPI),合并后可以看到哪些细胞同时表达Oct3/4和2A
读图细节
Ki67染色(Fig 1F):
- 问题:Dox处理后出现的异型增生细胞是不是真的在增殖?
- 方法:Ki67免疫组化(Ki67只在增殖细胞中表达)
- 答案:异型增生细胞Ki67阳性 → 确实在活跃增殖
Oct3/4 + 2A双重免疫荧光(Fig 1G):
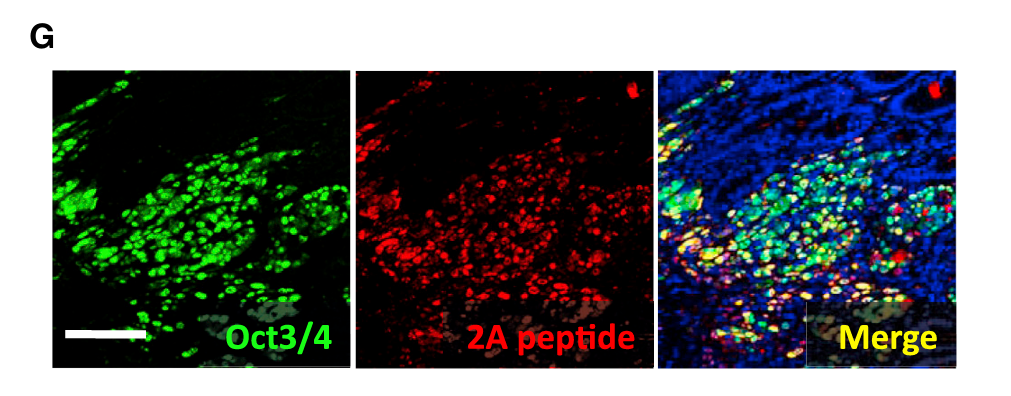
- 问题:那些增殖的异型细胞是不是因为表达了我们装进去的转基因?
- 方法:同时检测Oct3/4(重编程因子之一)和2A肽(转基因特有的连接序列)
- 答案:异型细胞同时表达两者 → 确认是转基因在驱动这些细胞的变化
2A染色(Fig S2B,论文文字描述):
- 问题:Dox撤除后长出来的肿瘤,还在表达转基因吗?
- 方法:2A免疫染色
- 答案:2A阴性 → 肿瘤生长已经不依赖转基因了 → 这非常关键,说明肿瘤是"自主性"的
Insulin + BrdU双重免疫荧光(Fig 2B):
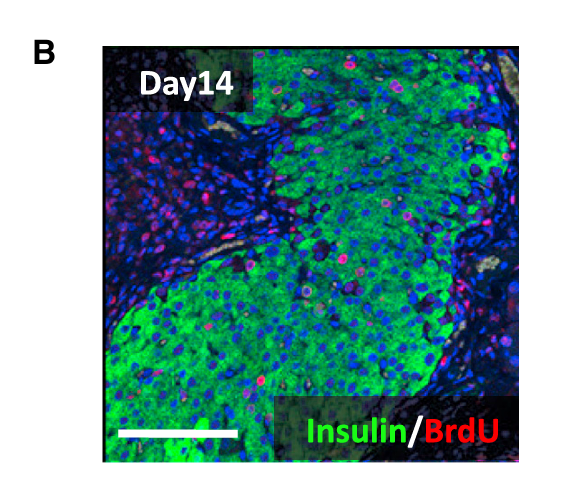
- 问题:短暂重编程后增殖过的细胞,撤药后去了哪里?变成了什么?
- 方法:Dox期间注射BrdU标记增殖细胞 → 撤药后同时检测BrdU(标记曾经增殖的细胞)和Insulin(胰岛β细胞标记)
- 答案:BrdU和Insulin在同一个细胞里共表达 → 说明曾经因重编程而增殖的细胞,撤药后变回了能分泌胰岛素的正常β细胞
GFP(Lgr5)+ Lin28b / Oct3/4 双重免疫荧光(Fig 3F):
- 问题:肿瘤特异性的Lgr5阳性细胞,有没有同时表达多能性相关蛋白?
- 方法:利用Lgr5-EGFP报告小鼠,GFP标记肿瘤细胞,同时检测Lin28b或Oct3/4
- 答案:部分GFP阳性细胞同时表达Lin28b和Oct3/4 → 肿瘤细胞处于"部分重编程"状态
方法3:FACS(流式细胞分选)
❤这一部分可以链接笔记流式细胞学部分的内容
原理:
细胞悬液 → 单细胞液流 → 激光照射 → 检测荧光信号 → 分选- 每个细胞通过激光时,仪器检测它发出的荧光(比如GFP绿色荧光)
- 根据荧光信号,仪器给每个细胞一个正或负的电荷
- 带电的细胞经过电场时被偏转到不同的收集管
在本论文中的用途:
- Fig 6A:从肾脏肿瘤中分选出Lgr5-GFP阳性细胞(肿瘤细胞)。因为肿瘤组织里不只有肿瘤细胞,还混有正常细胞、血管细胞、免疫细胞等。FACS可以把你想要的那群细胞"纯化"出来。
- mCherry分选:在Dox处理第7天,分选出mCherry阳性的肾脏细胞(即正在表达转基因的细胞),用于后续基因表达分析
探究基因表达模式的变化
方法1:qRT-PCR(查"单个基因"的表达量)
❤这一部分可以链接笔记qRT-PCR部分的内容
Fig 3A(Aqp1、Lrp2)、3C(Nanog、Lin28a、Esrrb)、3D(Lgr5)、4B(Six2)、5B(Igf2)、6B(Nanog)
原理:
第一步:RT(逆转录)——把mRNA变成DNA
mRNA → [逆转录酶] → cDNA(稳定,可以用PCR扩增)
第二步:qPCR(定量PCR)——测cDNA有多少Fig 3A——查肾脏身份有没有丢失:
- 检测Aqp1和Lrp2(都是肾小管细胞的特异标志基因)
- 结果:在肿瘤中显著下调 → 肿瘤细胞已经"忘了"自己是肾脏细胞
Fig 3C——查有没有获得干细胞特征:
- 检测Nanog、Lin28a(多能性标志)和Esrrb
- 结果:Nanog和Lin28a上调(但远低于ESC水平),Esrrb不变 → 部分获得了干性,但不完全
Fig 6B——查肿瘤细胞重编程到iPSC的速度:
- 分选Lgr5阳性肿瘤细胞,Dox处理7天后检测Nanog
- 结果:Nanog快速上升到接近ESC水平 → 说明肿瘤细胞比正常体细胞更容易被重编程(因为它已经"走了半程"了)
方法2:微阵列芯片(Microarray)——一次查"全部基因"
Fig 3B(ESC Core module热图)、4A(ESC PRC module热图)、4E、5D
原理:
微阵列芯片是一块玻璃片(大约指甲大小),上面按网格排列了成千上万个探针(probes)——每个探针是一段已知序列的短DNA,对应一个特定基因。
组织样本 → 提取RNA → 逆转录成cDNA → 标记荧光 → 放到芯片上杂交
芯片上的探针(已知基因A的序列)
↓ 如果样本中基因A有表达,其cDNA会结合上去
荧光信号强 → 基因A高表达
芯片上的探针(已知基因B的序列)
↓ 如果样本中基因B没有表达,没有cDNA能结合
荧光信号弱/无 → 基因B不表达扫描整块芯片,就能得到所有基因的表达量数据。
ESC基因表达三模块(来自Kim et al., Cell 2010):
之前的研究发现,ESC中高表达的基因可以分为三个功能组:
| 模块 | 包含的基因(举例) | 在ESC中的功能 |
|---|---|---|
| Core模块 | Nanog、Oct3/4、Sox2等 | 维持多能性的核心网络 |
| Myc模块 | c-Myc的靶基因们 | 促进增殖和代谢 |
| PRC模块 | 发育相关基因(Six2、Lgr5等) | 被Polycomb复合体沉默,等待分化时激活 |
肿瘤 vs ESC的对比:
Core模块:肿瘤 ≈ ESC(都激活)→ 肿瘤获得了自我更新能力 ✓
Myc模块:肿瘤 ≈ ESC(都激活)→ 肿瘤获得了增殖能力 ✓
PRC模块:肿瘤 ≠ ESC(肿瘤中未被沉默)→ 重编程"没做完" ✗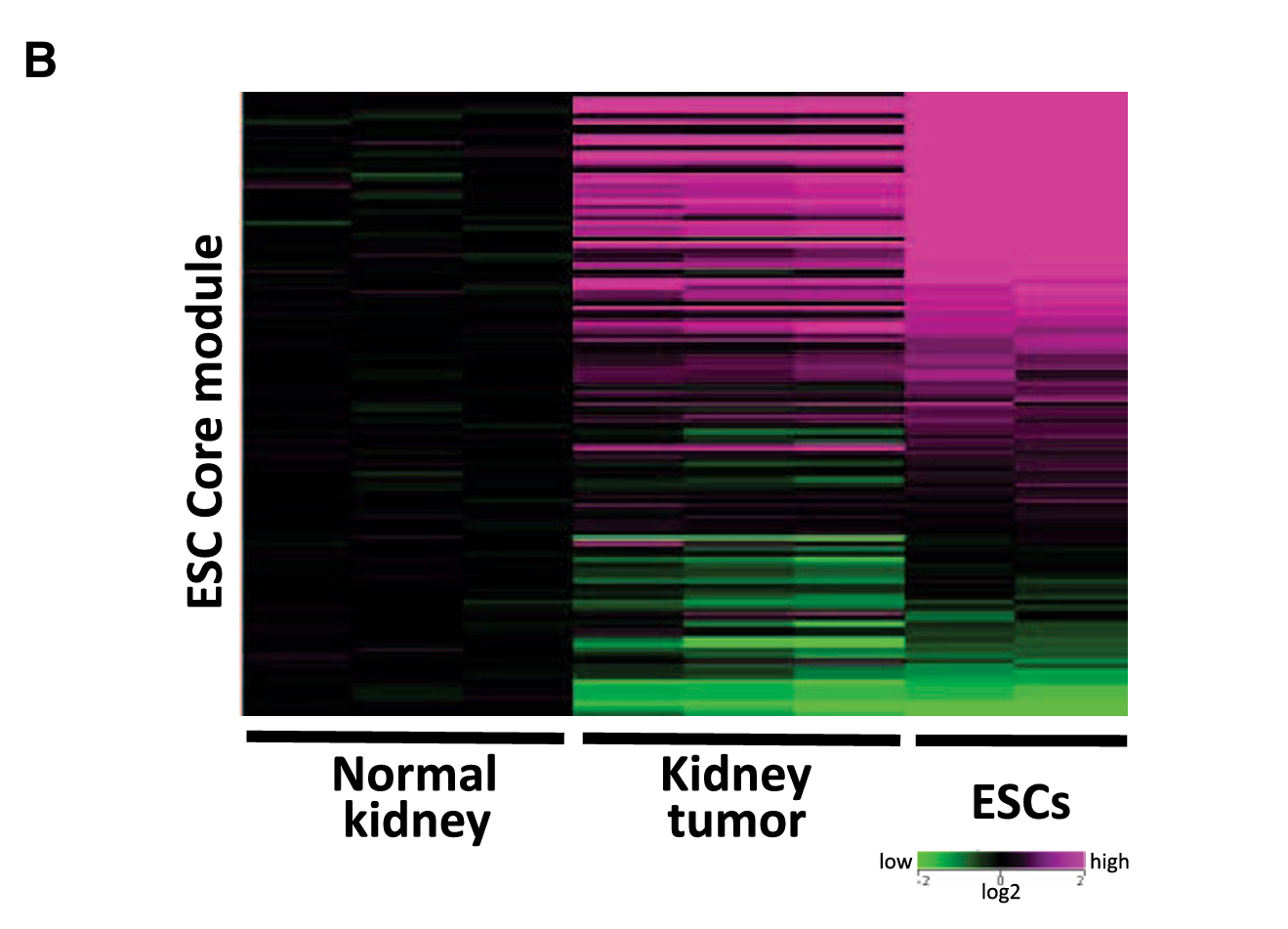
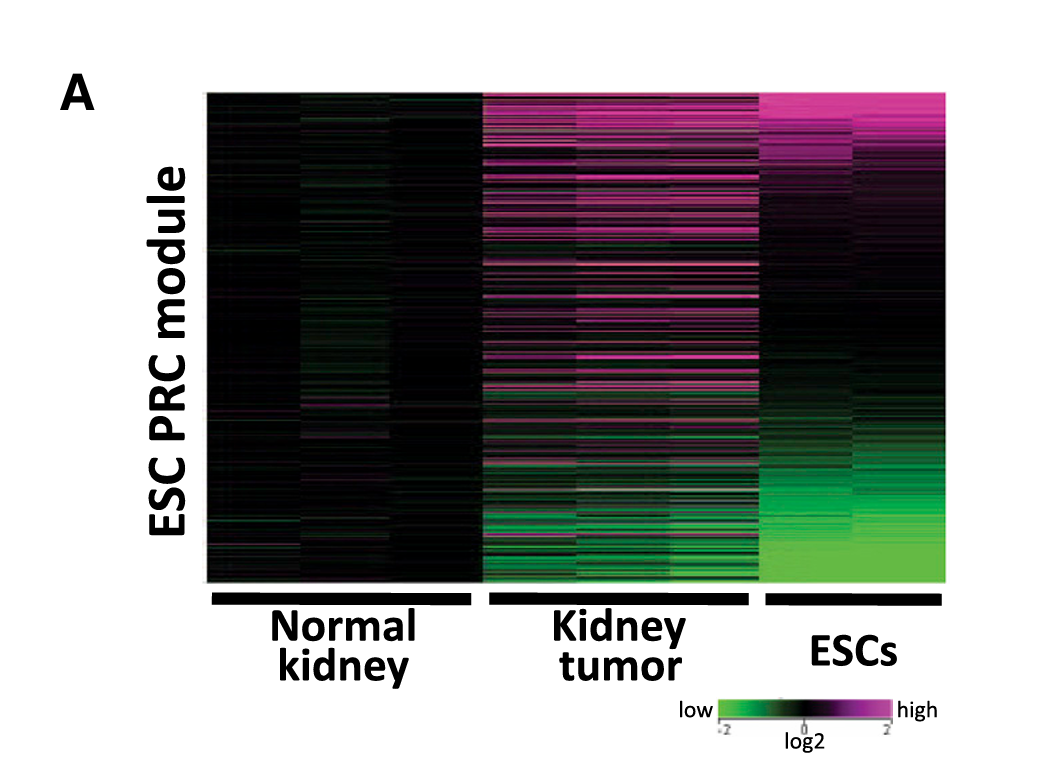
- 每一行代表一个基因
- 每一列代表一个样本(正常肾脏、肾脏肿瘤、ESC)
- 颜色代表表达量:红色=高表达、绿色/蓝色=低表达
- 如果两列的颜色模式很像,说明这两个样本的基因表达模式相似
Fig 3B:ESC Core module的热图中,肾脏肿瘤那一列的颜色模式和ESC很像(都偏红),而和正常肾脏很不同 → 说明肿瘤在Core模块层面已经像ESC了
探究表观遗传状态
甲基化:
未甲基化: ...C-G... → 基因通常可以表达(开关开着)
甲基化: ...ᵐC-G... → 基因通常被沉默(开关被锁上了)印记基因:
每个基因有两个拷贝——一个来自父亲,一个来自母亲,两个都表达。但有一小部分基因(约100个)很特殊:只从父方或母方的那个拷贝表达,另一个被甲基化沉默。 这种现象叫基因组印记(genomic imprinting)。控制印记的关键区域叫DMR(差异甲基化区域)——在这个区域,父方和母方的等位基因有不同的甲基化状态。
正常: 父方拷贝 → 甲基化(沉默)
母方拷贝 → 未甲基化(表达)
结果:只有母方等位基因表达
异常: 两个拷贝都去甲基化 → 双等位基因表达(表达量翻倍)
或: 两个拷贝都甲基化 → 完全沉默- 而重编程过程中,印记基因的甲基化状态会被打乱——所以印记异常是"不完全重编程"的标记
- 人类Wilms瘤最经典的分子改变就是Igf2/H19印记基因的甲基化异常——如果小鼠肿瘤也有这个改变,就能把小鼠模型和人类疾病联系起来
方法1:RRBS(查全基因组的甲基化状态)
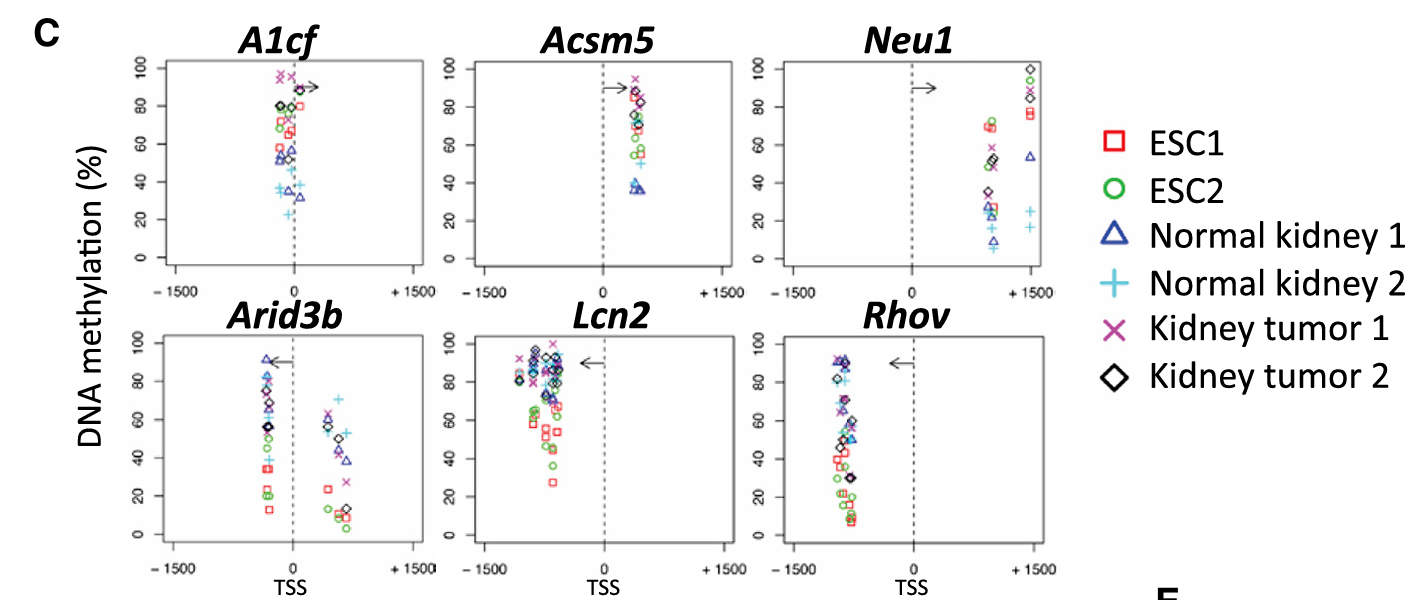
Fig 4C——单个基因层面的甲基化变化:
每个小图显示一个基因启动子区域(TSS±1500bp)的甲基化水平:
- X轴:距离转录起始位点(TSS)的距离
- Y轴:甲基化百分比(0-100%)
- 不同符号代表不同样本(ESC、正常肾脏、肾脏肿瘤)
例如A1cf基因:在ESC中高甲基化,在正常肾脏中低甲基化,在肿瘤中变成了高甲基化(像ESC) → 肿瘤获得了ESC样的甲基化模式。
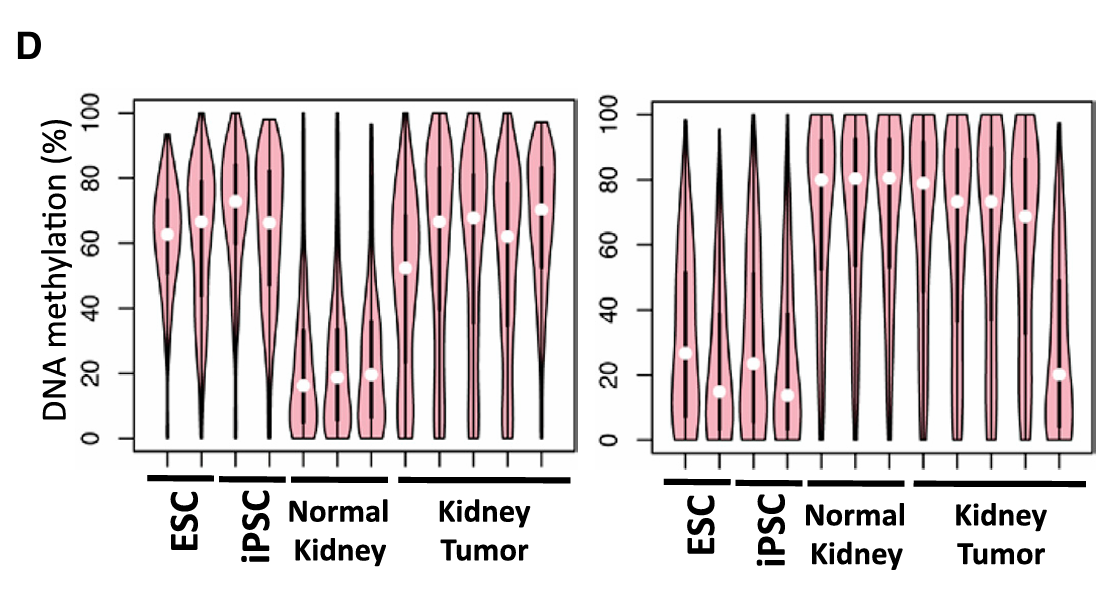
Fig 4D——全局性分析(最重要的图之一):
研究者做了一个非常聪明的分析:
- 先找出ESC和正常肾脏之间甲基化差异超过30%的基因
- 把这些基因分成两组:"ESC中甲基化的基因"和"肾脏中甲基化的基因"
- 分别看这两组基因在肿瘤中的甲基化状态
结果:
ESC中甲基化的基因 → 在肿瘤中也变成了高甲基化(像ESC)✓
肾脏中甲基化的基因 → 在肿瘤中仍然保持高甲基化(像肾脏)✓这意味着肿瘤细胞在重编程过程中先获得了ESC的甲基化模式,但还没来得及擦除肾脏细胞的甲基化模式——又一个"卡在半路"的证据。重编程进行到一半就被终止了。
方法2:亚硫酸氢盐测序(Bisulfite Sequencing)——查单个位点的精细状态
方法3:CGH和外显子测序——排除基因突变
这两个方法的目的不是"发现什么",而是排除什么——排除肿瘤是由基因突变驱动的可能性。
CGH(比较基因组杂交):
- 查的是:染色体有没有大片段的缺失或扩增
- 结果:没有发现显著的染色体异常
外显子测序(514个癌症相关基因):
- 查的是:已知的癌症驱动基因有没有突变(包括Wilms瘤相关的Wt1、Wtx、Ctnnb1、Trp53)
- 结果:没有发现癌症相关基因突变
证据链:
表观遗传确实发生了改变 → RRBS和亚硫酸氢盐测序证明 ✓ 基因没有发生不可逆突变 → CGH和外显子测序证明 ✓ 表观遗传改变可以被逆转 → 肿瘤iPSC分化为正常组织证明 ✓
结论:表观遗传异常独立驱动了癌症发生
总结
- 基因工程造模 → 制造可遥控的Reprogrammable小鼠
- 组织学+免疫染色 → 看到肿瘤形成,确认细胞类型和增殖状态
- 基因表达分析 → 发现肿瘤的基因表达是"卡在重编程中间"的状态
- 表观遗传分析 → 证明DNA甲基化确实被改变了,而DNA序列没有突变
- 所有证据汇聚 → 表观遗传异常可以独立驱动癌症
关键发现
表观遗传异常可以独立驱动癌症
与研究室其他工作的关系
山田研究室重编程方向的基石性文章
面试可能被问到的问题
准备回答
- 你对这篇论文的方法有什么看法?
- 你觉得这个方法可以怎么改进?
- 这个方向未来的发展趋势是什么?
个人思考
这一篇文章很重磅,发在了Cell上面,所以精读的比较久,查阅了很多资料。
早在2005年,Yasihiro Yamada教授在Jaenisch实验室做博后时,就和Hochedlinger共同发表了一篇Cell论文,发现在成体小鼠中强制表达Oct3/4会导致上皮组织的异型增生。但那篇论文中,撤除Oct3/4后异型增生完全消失了——没有形成真正的肿瘤。所以那只是说明Oct3/4可以阻断分化,但没有证明重编程可以独立致癌。同时期也有其他研究者注意到iPSC重编程和癌症共享很多分子通路(比如p53通路的抑制对两者都是促进的)。但这些都是"相关性",不是"因果性"。
在这篇文章之前,很多学者观察到细胞会"重编程失败",停在中间状态——这些部分重编程细胞(partially reprogrammed cells)的基因表达和表观遗传确实有异常。但没有人系统地研究过这些细胞在活体内会不会变成癌症。大多只是在培养皿中验证。
且在这篇论文之前,科学界已经充分认识到癌细胞中普遍存在表观遗传异常,比如启动子区CpG岛的高甲基化导致抑癌基因沉默、全基因组的低甲基化导致基因组不稳定等。但长期以来存在一个根本性的争论:到底是基因突变先发生导致了表观遗传异常还是表观遗传异常先发生独立驱动了癌变亦或者是两者共同作用。多数人倾向于认为基因突变是主要驱动力,表观遗传异常只是伴随现象或次要因素。原因很简单:没有人能在不引入基因突变的情况下,仅靠改变表观遗传就在活体内造出真正的癌症。
而Yasihiro Yamada教授的这篇文章就建立了体内可控的"不完全重编程"系统。 通过Tet-on系统精确控制Dox给药天数,可以在活体内制造"重编程中断"这个状态。并且证明了不完全重编程在体内产生的是真正的恶性肿瘤,而非良性增生。还提供了"表观遗传驱动、非基因突变驱动"的完整证据链。
① 重编程本身不改变DNA序列(这是iPSC技术的已知前提)
② 不完全重编程导致了肿瘤(Dox实验直接观察到)
③ 肿瘤中确实存在大量表观遗传改变(RRBS证明)
④ 肿瘤中找不到癌症驱动基因突变(外显子测序+CGH证明)
⑤ 肿瘤来源的iPSC可以分化为正常组织且不再致癌(终极验证)叹为观止!