Appearance
Wnt/β-catenin信号通路通过抑制CEBPA和FOXA1调控肝癌细胞氨基酸代谢(2024)
论文信息
教授: Yoichi Furukawa · 大学: 东京大学医科学研究所 · 阅读日期: 2026-05-10 ~ 2026-05-17
基本信息
| 项目 | 内容 |
|---|---|
| 标题 | Wnt/β-catenin signaling regulates amino acid metabolism through the suppression of CEBPA and FOXA1 in liver cancer cells |
| 作者 | Saya Nakagawa, Kiyoshi Yamaguchi, Kiyoko Takane, Sho Tabata, Tsuneo Ikenoue, Yoichi Furukawa |
| 发表于 | Communications Biology (2024) 7:510 |
| DOI/链接 | 10.1038/s42003-024-06202-9 |
| 被引次数 | - |
总结概要
在肝癌细胞中,Wnt/β-catenin信号通路被异常激活后会间接抑制两个肝脏富集的转录因子CEBPA和FOXA1,进而下调它们调控的代谢酶HAL(组氨酸氨裂解酶)和ARG1(精氨酸酶1)。这导致肝癌细胞内组氨酸和精氨酸累积,尿素循环被抑制,同时乳酸水平上升(Warburg效应)。这是首次系统地把Wnt信号 → 肝特异性转录因子下调 → 氨基酸代谢重编程这条轴连起来的工作。
研究背景与动机
- Wnt/β-catenin通路异常激活是消化器癌(尤其是大肠癌和肝癌)的核心驱动事件。HCC中约31%携带CTNNB1突变、6%携带AXIN1突变,导致β-catenin入核与TCF/LEF结合,激活下游靶基因
- 经典研究多关注β-catenin/TCF复合体正向激活的靶基因(如c-Myc、Cyclin D1、AXIN2、LGR5等),但被Wnt信号下调的基因长期被忽视
- 古川研究室此前已发表过《Biotechnol Bioeng 2017》,建立了HAL启动子 + TOPFLASH双向报告基因系统,并发现HAL在肝癌中被Wnt信号转录性抑制。但是——β-catenin/TCF复合体本身是转录激活因子,怎么会"抑制"HAL? 一定是中间存在某个"中介转录因子"。这个中介因子是谁?这就是本研究要回答的问题
- 同时,Wnt信号与糖代谢(Warburg效应)的联系已较为清楚(MYC、PDK1、MCT1等),但与氨基酸代谢的关系几乎是空白。本研究试图填补这块空白
核心方法
整体思路
找中介 → 验证中介 → 找下游 → 看代谢后果
- 找候选转录因子:用JASPAR数据库扫描HAL启动子-90/-44bp区域的TF结合位点,结合微阵列芯片筛选受Wnt信号调控的TF
- 验证转录调控:报告基因检测 + siRNA敲低 + 过表达 + 突变位点
- 找下游靶基因:RNA-seq(找差异表达基因)+ ChIP-seq(找直接结合位点)→ 取交集
- 代谢后果:CE-TOFMS代谢组学分析
实验流程图
HAL启动子-90/-44bp区域
↓ JASPAR扫描TF结合位点 + 微阵列筛Wnt靶基因
33个候选TF
↓ 与HAL表达相关性分析(TCGA)+ siRNA验证
CEBPA + FOXA1(中介转录因子)
↓ RNA-seq + ChIP-seq 求交集
132个共同直接靶基因
↓ KEGG通路富集
"精氨酸和脯氨酸代谢" → ARG1
↓ 代谢组学(CE-TOFMS)
组氨酸↑、精氨酸↑、乳酸↑、尿素循环紊乱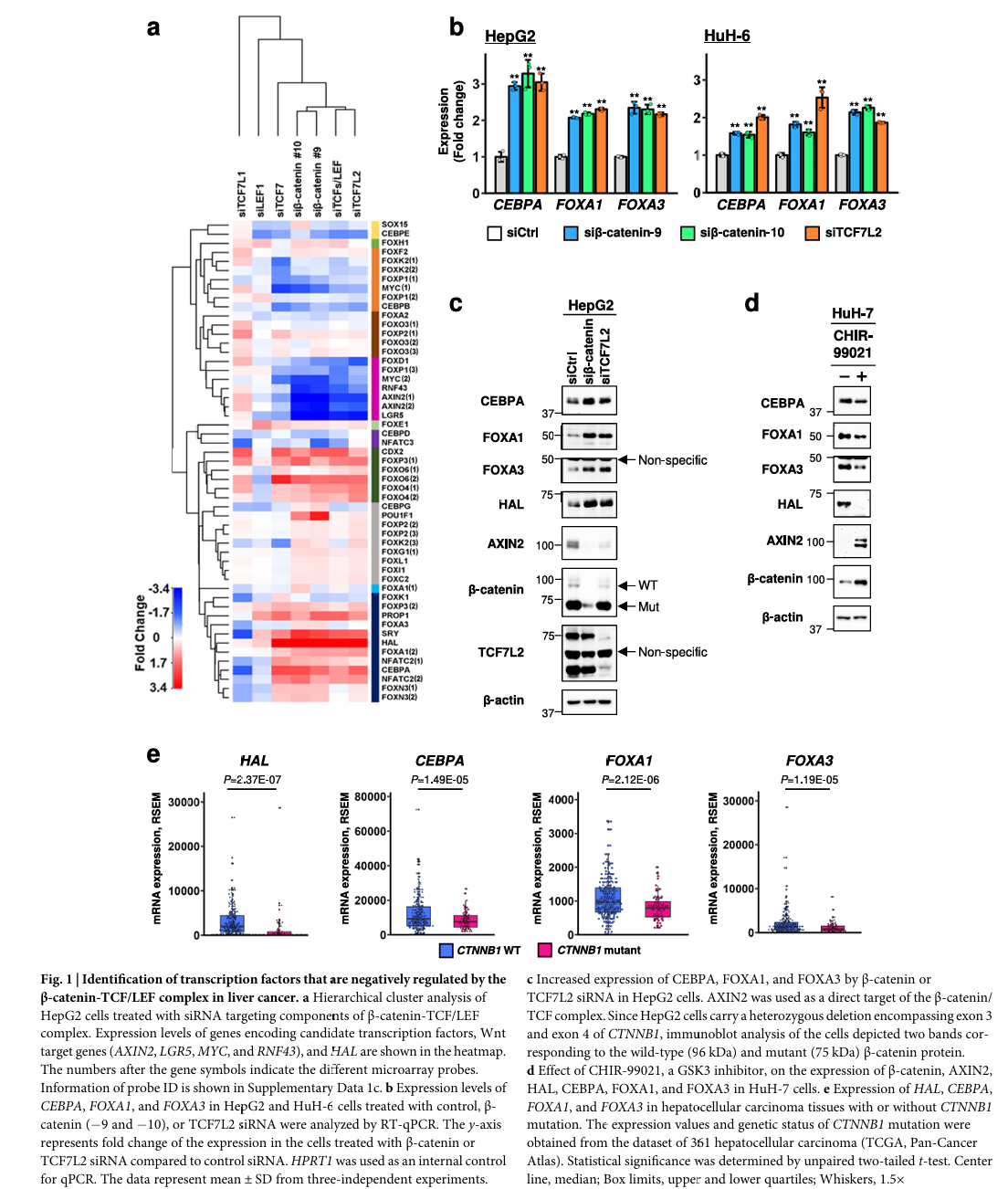
实验与结果
第一部分:寻找调控HAL的转录因子
方法1:JASPAR数据库扫描 + 微阵列筛选
❤这一部分可以链接笔记[Yoichi Furukawa论文(1)补充知识点](/notes/Yoichi Furukawa论文(1)补充知识点)部分的内容
JASPAR是一个公开的转录因子结合谱(TF binding profile)数据库,存储了上千个TF的位置权重矩阵(PWM)。给一段DNA序列,可以扫描出可能结合的TF。
原理:
HAL启动子-90/-44bp区域(已知是Wnt信号"间接"作用的关键区域)
↓ JASPAR扫描(score ≥ 8.0)
33个候选转录因子
↓ 把这33个TF的表达量放进微阵列结果里看
看哪些TF被β-catenin/TCF7L2 siRNA改变了表达关键发现(Fig 1a 热图):
- 通过层次聚类分析,发现FOXD1、FOXP1与已知Wnt靶基因(MYC、RNF43、AXIN2、LGR5)聚成一类——它们被Wnt正向激活
- CEBPA、FOXA1、FOXA3等9个TF与HAL聚成一类——它们与HAL同步变化,且都被β-catenin/TCF7L2 siRNA上调
- 这9个TF就是候选激活因子——可能是它们激活了HAL,而Wnt信号通过抑制它们间接抑制HAL
方法2:用TCGA数据筛选最相关的候选
把9个候选TF放进TCGA肝癌数据集(cBioPortal),看哪些TF的表达水平与HAL在患者样本中显著正相关:
| 候选TF | 与HAL的相关系数(r) | q值 |
|---|---|---|
| FOXA3 | 0.36 | < 0.01 |
| FOXA1 | 0.25 | < 0.01 |
| CEBPA | 0.22 | < 0.01 |
三个候选锁定:CEBPA、FOXA1、FOXA3——而这三个都是肝脏富集的转录因子(liver-enriched TFs),与HAL一起在正常肝组织中高表达。
方法3:qPCR + 免疫印迹 + 临床数据交叉验证
Fig 1b、c:在HepG2和HuH-6细胞(两种都携带CTNNB1激活突变)中,敲低β-catenin或TCF7L2后,三个TF的mRNA和蛋白都上调
Fig 1d:反向验证——用GSK-3抑制剂CHIR-99021激活Wnt信号,三个TF表达下降
Fig 1e:TCGA中CTNNB1突变型HCC比野生型HCC的HAL、CEBPA、FOXA1、FOXA3表达都显著降低(p < 1e-5)
读图细节
Fig 1a 热图:
- 行:候选TF + Wnt靶基因 + HAL
- 列:不同siRNA处理组(siTCF7L1、siLEF1、siTCF7、siβ-catenin#10、siβ-catenin#9、siTCF/LEF、siTCF7L2)
- 红色 = 上调(敲低Wnt组分后表达升高),蓝色 = 下调
- 关键观察:HAL那行明显被红色框圈出来——siβ-catenin和siTCF7L2两列都是深红色,证明Wnt信号抑制HAL。同时CEBPA、FOXA1、FOXA3和HAL在热图上"长得很像",说明它们被同步调控
注意HepG2细胞的特殊性: 它带有CTNNB1外显子3和4的杂合性缺失(删去了磷酸化降解信号),所以免疫印迹时会看到两条带:96 kDa(野生型β-catenin)和75 kDa(突变型β-catenin)。这是肝癌细胞模型选择的常见考量——选已经激活Wnt通路的细胞系。
第二部分:验证CEBPA和FOXA1是HAL的直接调控因子
方法1:报告基因实验 + 位点定向突变
原理:
HAL启动子-90/+147区域 → 萤火虫荧光素酶基因 (pHAL-90/+147)
↓
转染入细胞
↓
测荧光强度
↓
反映启动子活性研究者在-90/-44bp区域内识别出3个潜在结合位点:
- CBE-1(CEBP结合元件1)
- CBE-2(CEBP结合元件2)
- FBE(Forkhead结合元件,FOXA1/3可结合)
然后做了位点定向突变(site-directed mutagenesis)——把每个结合位点的关键碱基突变掉,看启动子活性变化(Fig 2a):
WT (野生型) → 完整功能
CBE-1m (CBE-1突变) → 部分丧失
CBE-2m (CBE-2突变) → 部分丧失
FBEm (FBE突变) → 部分丧失
CBE-1m+CBE-2m+FBEm (三重突变) → 几乎完全丧失Fig 2b:si-β-catenin处理后HAL启动子活性上升7倍,但三重突变后这种上升几乎消失 → 证明这三个位点确实是Wnt信号"间接抑制"HAL的关键
Fig 2c:过表达CEBPA显著增加HAL启动子活性(增加约20倍),但单独过表达FOXA1或FOXA3没什么效果
Fig 2d:敲低CEBPA或FOXA1都能降低HAL启动子活性,但FOXA3敲低不影响
关于FOXA1的"前导因子"角色
FOXA1是一个先驱因子(pioneer factor)——这类TF的特殊之处在于它们可以识别包装在核小体上的DNA序列,并打开染色质结构,让其他TF能够进入。
这解释了为什么实验中存在一个"矛盾":
- 报告基因实验:FOXA1单独过表达对HAL启动子活性影响不大(Fig 2c)
- 细胞实验:FOXA1敲低能显著降低内源性HAL表达(Fig 3a)
为什么矛盾? 因为报告基因质粒上的DNA是裸露的,不需要先驱因子来打开染色质。而细胞内基因组DNA是包装在核小体上的,需要FOXA1先打开染色质,CEBPA才能结合上去发挥转录激活作用。
方法2:qPCR + 免疫印迹(在HuH-7和Hep3B两种细胞中验证)
Fig 3a, b:敲低CEBPA或FOXA1,HAL的mRNA和蛋白都显著下降 Fig 3c, d:过表达CEBPA或FOXA1,HAL显著上升
关键阴性对照:FOXA3虽然在TCGA中与HAL相关性最高(r=0.36),但功能实验中过表达不能诱导HAL、敲低也不能稳定降低HAL → 说明相关性≠因果性,TCGA的相关性可能反映FOXA3和HAL都被同一个上游因素调控(比如肝脏分化程度),但FOXA3本身不直接调控HAL
第三部分:找CEBPA和FOXA1的下游靶基因
方法1:RNA-seq + ChIP-seq 求交集
思路:
- RNA-seq告诉你"哪些基因因为TF的存在/缺失而表达变化"——但变化可能是直接的(TF直接结合)或间接的(TF调控了别的因子再调控这个基因)
- ChIP-seq告诉你"TF在基因组上的实际结合位点"——但结合不一定意味着调控
- 两者交集就是直接靶基因——既被TF结合,又因TF敲低而下调
RNA-seq (siCEBPA下调的基因) ∩ ChIP-seq (CEBPA结合的基因) = 460个CEBPA直接靶基因
RNA-seq (siFOXA1下调的基因) ∩ ChIP-seq (FOXA1结合的基因) = 489个FOXA1直接靶基因
两者再求交集 = 132个CEBPA和FOXA1共同的直接靶基因(包含HAL)Fig 4a:在HAL基因5'侧翼区域可以清楚看到CEBPA(紫红色)和FOXA1(蓝色)的ChIP-seq峰
Fig 4b:维恩图,分别显示CEBPA和FOXA1的直接靶基因数量
方法2:KEGG通路富集分析
❤这一部分可以链接笔记基因集富集分析部分的内容
把132个共同靶基因送进KEGG数据库富集分析(Fig 4e):
最显著富集的通路:精氨酸和脯氨酸代谢(Arginine and proline metabolism)
这个通路里有4个差异表达基因:AMD1、ARG1、GLS、GOT1。
方法3:筛选出真正受Wnt信号调控的下游
Fig 4f:在HepG2细胞中,用si-β-catenin和si-TCF7L2检测这4个基因。结果只有ARG1显著上调(敲低Wnt信号后ARG1上升约20倍),其他三个基因要么下调要么变化不大。
Fig 4g:ARG1基因的内含子1中有清晰的CEBPA和FOXA1的ChIP-seq结合峰
Fig 4h(最关键的因果实验):
si-Ctrl: HAL低, ARG1低
si-β-catenin单独: HAL高, ARG1高 ← 敲低Wnt → 解除抑制
si-β-catenin + si-CEBPA: HAL中, ARG1中 ← 加回CEBPA敲低 → 又抑制了
si-β-catenin + si-FOXA1: HAL中, ARG1中 ← 加回FOXA1敲低 → 又抑制了这种"双敲回补实验(epistasis experiment)"是这篇论文的金标准——证明了Wnt → CEBPA/FOXA1 → HAL/ARG1这条线性通路。
第四部分:代谢组学验证
方法:CE-TOFMS代谢组学
**CE-TOFMS(毛细管电泳-飞行时间质谱)**原理:
细胞裂解液 → 加入内参 → 过5kDa超滤膜(去蛋白)
↓
毛细管电泳(按带电性质分离化合物)
↓
飞行时间质谱(按质荷比m/z鉴定)
↓
比对Human Metabolome Technologies数据库 → 鉴定出116种代谢物CE-TOFMS的优势在于对小分子带电代谢物(氨基酸、有机酸、核苷酸)分离效果极好——正好适合本研究。
Fig 5a 热图:116种代谢物中,β-catenin和TCF7L2 siRNA共同改变的代谢物大多是下降的
Fig 5b:精氨酸和组氨酸在si-β-catenin和si-TCF7L2组都显著降低(这正是HAL和ARG1上调把它们消耗掉的结果)
Fig 5c:
- 乳酸下降 → 提示Wnt信号通路抑制后,糖酵解减弱(Warburg效应减弱)
- 乙酰辅酶A上升 → 提示丙酮酸更多进入TCA循环而非生成乳酸
Fig 5d 通路富集分析:富集最显著的代谢通路是尿素循环(Urea Cycle)——因为ARG1是尿素循环的核心酶之一
Fig 5e:进一步检查尿素循环其他酶基因(OTC、ASS1、ASL)的表达,发现这些基因在敲低Wnt信号后都显著上升 → 说明激活的Wnt信号系统性地抑制了整个尿素循环
致癌生物学意义的解读
Wnt激活
↓
CEBPA/FOXA1抑制
↓
ARG1抑制 → 尿素循环抑制 → 氨基酸不被分解→保留作为蛋白合成原料 → 帮助快速增殖
+
HAL抑制 → 组氨酸保留 → 通过四氢叶酸途径 → 嘌呤合成 → 帮助DNA复制 + 化疗耐药(甲氨蝶呤)
+
PDK1激活 → Warburg效应 → 乳酸累积 → 免疫逃逸关键发现
Wnt/β-catenin信号通路在肝癌中不仅是经典的"促增殖信号",更是一个"代谢重编程指挥棒"——它通过下调肝特异性转录因子CEBPA和FOXA1,系统性地重塑肝癌细胞的氨基酸代谢和尿素循环,为癌细胞快速增殖提供代谢支持,同时可能促进免疫逃逸和化疗耐药。
与研究室其他工作的关系
这篇是古川研究室Wnt信号研究主线上的最新延伸:
| 时间 | 工作 | 内容 |
|---|---|---|
| 2000 | Satoh et al., Nature Genet | 首次报道AXIN1突变在HCC中存在 |
| 2002 | Takahashi et al., Cancer Res | 鉴定APCDD1为Wnt直接靶基因 |
| 2011 | Yamaguchi et al., Cancer Sci | 发现MRGBP是β-catenin/TCF下游 |
| 2017 | Yamaguchi et al., Biotechnol Bioeng | 建立HAL启动子 + TOPFLASH双向报告系统,发现HAL被Wnt抑制 |
| 2019 | Ohsugi et al., Oncogene | IRF1抑制在大肠癌中的作用 |
| 2023 | Yamaguchi et al., iScience | BRD8调控大肠癌细胞周期 |
| 2024 | Nakagawa et al., Commun Biol | 本文:揭示Wnt→CEBPA/FOXA1→氨基酸代谢轴 |
本文回答了2017年那篇"HAL被Wnt抑制"留下的最大悬念:抑制机制是什么? 同时把研究室的方向从"基因表达调控"扩展到"代谢重编程",与目前肿瘤代谢研究的全球趋势接轨。
对抗式审阅
一、论证链条中最薄弱的环节
漏洞1:因果链条上的"间接性"——多个关键步骤都是相关性而非因果性证明
虽然作者声称建立了Wnt → CEBPA/FOXA1 → HAL/ARG1 → 氨基酸代谢这条线性通路,但仔细审视会发现:
问题①:β-catenin/TCF如何抑制CEBPA和FOXA1的转录? 论文完全没有说明机制。
- 没有做CEBPA和FOXA1启动子的ChIP-seq(看β-catenin/TCF7L2是否直接结合)
- 没有做CEBPA和FOXA1启动子的报告基因实验
- 在讨论部分作者承认"在结肠癌细胞中β-catenin敲低不能诱导CEBPA和FOXA1"——这暗示存在某个"肝特异性的中介因子",但作者完全没有去寻找它
这个漏洞如何影响结论可信度:如果β-catenin对CEBPA和FOXA1的抑制是通过某个未知的中介因子完成的,那么这条通路就不是"Wnt → CEBPA/FOXA1"两步,而是"Wnt → X → CEBPA/FOXA1"三步。X是什么?X在不同癌型中是否存在差异?X本身是否就是真正的治疗靶点?这些都是悬而未决的。
问题②:双敲回补实验(Fig 4h)的解读问题。 这个实验是论文的核心,但仔细看图:
- siβ-catenin + siCEBPA组合中,HAL和ARG1的"回降"是部分的,不是完全的
- 这说明CEBPA和FOXA1并不能完全解释Wnt信号对HAL/ARG1的调控,可能还有其他中介因子并行存在(作者在讨论中提到HNF4A也调控HAL,但没有完整整合进通路)
漏洞2:研究对象的局限——主要在Wnt已经激活的细胞系中验证
所有四个肝癌细胞系(HepG2、HuH-6、HuH-7、Hep3B)的Wnt信号状态各不相同:
| 细胞系 | CTNNB1状态 | AXIN1状态 |
|---|---|---|
| HepG2 | 突变(外显子3-4缺失) | 野生 |
| HuH-6 | 突变 | 野生 |
| Hep3B | 野生 | 突变 |
| HuH-7 | 野生 | 野生 |
问题:HuH-7是Wnt信号相对静息的细胞,但论文中RNA-seq和ChIP-seq关键实验都在HuH-7中进行。这意味着作者在Wnt信号弱的细胞中找CEBPA/FOXA1靶基因,然后回到Wnt信号强的细胞中验证表型——这种"系统不一致"在生物学上可能引入偏差。
更严格的做法是:使用同一对等基因细胞系(isogenic cell lines)——比如CRISPR-Cas9敲入或修复CTNNB1突变,从而把"Wnt信号差异"作为唯一变量。
漏洞3:体内证据严重缺失
这是最致命的漏洞——全篇论文没有任何体内(in vivo)实验。
- 没有异种移植瘤(xenograft)实验
- 没有肝特异性Apc或Ctnnb1突变小鼠模型的验证(虽然作者在讨论中引用了别人2009年的Apc-KO小鼠蛋白组学数据,但没有自己做)
- 没有患者来源类器官(PDO)的验证
这个漏洞如何影响结论可信度:所有代谢组学数据都是在HepG2细胞(培养条件下)做的。但培养基中的氨基酸浓度远远高于生理水平——HepG2培养基中精氨酸约0.4 mM,而正常肝细胞外液中精氨酸约0.08 mM。在外源精氨酸过剩的情况下观察到的"精氨酸累积",是否能外推到体内环境?这是一个严重的问题。
漏洞4:代谢组学的局限——只看了细胞内代谢物水平,没有看通量(flux)
代谢组学只是个"快照"——它告诉你某个时刻细胞里有多少代谢物,但不告诉你这些代谢物流动的速度。
精氨酸水平升高可以由两种完全相反的机制造成:
- 机制A:ARG1降低→精氨酸不被消耗→精氨酸累积
- 机制B:精氨酸合成加速→ARG1根本来不及消耗→精氨酸累积
要区分这两种机制,需要做同位素示踪代谢通量分析(¹³C-flux analysis)——比如喂细胞¹³C-精氨酸,追踪它进入尿素循环的速度。论文没有做这个实验,所以"Wnt信号抑制尿素循环"这个结论严格来说是推测性的。
漏洞5:临床相关性论证薄弱
论文使用了TCGA数据来支撑临床相关性,但是:
- 只看了表达水平的关联(mRNA水平的相关性),没有看蛋白水平
- 没有分析CEBPA/FOXA1或HAL/ARG1表达与患者生存期、复发率、化疗反应的关系
- 没有做免疫组织化学验证(IHC在临床HCC样本上证明CTNNB1突变型患者确实CEBPA/FOXA1/HAL/ARG1表达降低)
二、隐含的、未被检验的前提假设
假设1:HepG2细胞代表了所有Wnt驱动的肝癌
HepG2是**肝母细胞瘤(hepatoblastoma)**来源的细胞系,严格来说不是成人型肝细胞癌(HCC)。两者的基因表达谱、代谢特征、临床行为都有差异。如果这个假设不成立,那么本文的所有结论可能只适用于儿童肝母细胞瘤,而非占肝癌大多数的成人HCC。
假设2:CEBPA和FOXA1在肝癌中的功能是"肿瘤抑制"
论文暗示Wnt信号抑制CEBPA/FOXA1是"促癌"的——但CEBPA和FOXA1本身在肝癌中的角色是复杂的:
- FOXA1在ER+乳腺癌中是促癌因子
- CEBPA在AML中是经典的肿瘤抑制因子,但在肝脏中的功能存在争议
如果这个假设不成立——比如CEBPA/FOXA1的下调可能只是细胞去分化的结果而非原因——那么本文揭示的就不是"致癌机制"而是"分化标志",治疗意义会大打折扣。
假设3:氨基酸代谢重编程对肝癌细胞增殖是必需的
论文给出了一个"代谢重编程"的图景,但没有验证这种重编程对肝癌细胞表型的功能性贡献:
- 在HepG2细胞中过表达HAL或ARG1,能否抑制增殖?
- 限制培养基中精氨酸/组氨酸供给,对Wnt激活型vs Wnt静息型细胞的影响是否不同?
- 使用ARG1抑制剂(如nor-NOHA)或精氨酸耗竭剂(如pegylated arginase)治疗Wnt激活型HCC类器官,是否有效?
如果这个假设不成立——即氨基酸代谢重编程只是"伴随现象"而非"功能驱动"——那么本文的治疗潜力指向就会失去基础。
假设4:Wnt信号在体内对代谢的调控与体外一致
体外实验剥离了**肿瘤微环境(TME)**的影响。在体内:
- 肝癌肿瘤区域常有缺氧、营养限制
- 免疫细胞、星状细胞、内皮细胞构成的微环境会显著影响代谢
- 全身代谢状态(如肝硬化背景)也会改变肝癌的代谢
如果这个假设不成立——比如体内的肝癌细胞因为微环境压力而表现出与体外完全不同的代谢模式——那么本文揭示的"代谢轴"在体内可能根本不被激活。
三、论文回避讨论的重要问题
回避1:CEBPA/FOXA1下调的"机制黑箱"
如前所述,β-catenin是转录激活因子,怎么能"抑制"CEBPA/FOXA1的转录?论文完全回避了这个核心机制问题。可能性包括:
- β-catenin/TCF激活某个抑制性TF(已知例子如TRIB2)
- β-catenin影响染色质修饰(如组蛋白去乙酰化)
- 通过miRNA介导
- 通过竞争性结合共激活因子
不去讨论这个机制,本文的结论就停在"现象描述"层面,无法指向真正的治疗靶点。
回避2:FOXA3的"鸡肋"结果
FOXA3在TCGA相关性分析中是最高的(r=0.36),但功能实验完全阴性。作者只是简单地"排除"了FOXA3,但没有解释:
- 为什么相关性最高的TF反而没有功能?
- FOXA3是否调控其他Wnt下游代谢基因?
- FOXA3的下调是否是CEBPA/FOXA1下调的"结果"(cascade效应)?
回避3:Wnt信号的"双面性"被忽视
Wnt信号在肝脏有强烈的位置依赖性(zonation)——中央静脉周围的肝细胞(perivenous hepatocytes)本身就有较高的Wnt活性,并表达不同的代谢酶谱(如GS、GLT1等高表达,ARG1、HAL低表达)。这是生理状态,不是病理状态。
那么——
- 本文观察到的"Wnt → CEBPA/FOXA1↓ → HAL/ARG1↓"是否只是生理性肝脏分区程序被肿瘤"劫持"?
- CTNNB1突变型HCC是否表现为"perivenous-like"分化?
- 这种"分化方向偏移"是否可以通过促分化治疗逆转?
作者完全没有把这篇论文放进肝脏代谢分区的大背景里讨论,这是一个重大遗漏。
回避4:免疫调控的暗示但未深入
讨论中作者提到"乳酸累积可能促进免疫逃逸",但完全没有提供数据。这只是基于其他人工作的推测。
此外,ARG1本身在肿瘤免疫中是一把双刃剑:
- 肿瘤细胞内的ARG1降低→精氨酸保留→促进增殖(本文观点)
- 但髓系来源抑制细胞(MDSC)的ARG1升高→消耗微环境精氨酸→T细胞功能抑制(既有文献观点)
那么在Wnt激活型HCC的微环境中,到底是"肿瘤细胞内ARG1低"占主导还是"MDSC的ARG1高"占主导?这对免疫治疗策略选择至关重要,但论文完全没提。
四、如果我是审稿人,我会要求作者补充什么数据或分析?
必须补充(major revision)
CEBPA/FOXA1启动子区的ChIP-seq或ChIP-qPCR——证明β-catenin/TCF7L2是否直接结合到这两个基因的调控区域,从而区分"直接抑制"vs"间接抑制"
体内实验——至少要补充一个:
- 用CTNNB1突变型小鼠肝癌模型(如AAV-Cre诱导的肝特异性Apc敲除),看HAL和ARG1的表达
- 用CEBPA/FOXA1过表达的HepG2细胞做异种移植瘤,看是否抑制肿瘤生长
- 在PDX或PDO中验证Wnt信号与CEBPA/FOXA1/HAL/ARG1的关联
¹³C-同位素示踪代谢通量分析——用¹³C-精氨酸或¹³C-组氨酸喂细胞,定量分析这些氨基酸的代谢通量,区分"累积"和"通量降低"
功能性回补实验——
- 在Wnt激活的HepG2细胞中强制过表达HAL或ARG1,看是否抑制增殖
- 在Wnt激活的HepG2细胞中限制培养基精氨酸/组氨酸,看是否选择性杀伤
- 这些实验直接回答了"代谢重编程是否对肿瘤表型必需"
强烈建议补充(minor revision)
临床预后分析——TCGA HCC队列中按CEBPA/FOXA1/HAL/ARG1表达分组,看生存期、复发率、化疗反应
HCC组织微阵列的免疫组化——CTNNB1突变型vs野生型HCC的CEBPA/FOXA1/HAL/ARG1蛋白表达对比
更严格的细胞模型——用CRISPR在Wnt静息细胞中敲入CTNNB1激活突变,或在Wnt激活细胞中修复CTNNB1,作为isogenic对照
FOXA3的进一步探索——做FOXA3的ChIP-seq,看它在Wnt激活细胞和静息细胞中的结合谱差异
肝脏分区背景的讨论——把Wnt信号的生理性分区与肿瘤性激活进行系统对比
免疫微环境的探索——分析CTNNB1突变型HCC的TME特征(如CD8+ T细胞浸润、PD-L1表达),看是否与乳酸/精氨酸代谢相关
五、综合评价
这篇论文作为机制研究是可以接受的——它扎实地建立了Wnt→CEBPA/FOXA1→HAL/ARG1这条转录调控轴,方法学规范,逻辑链条完整。作为Communications Biology这个层级的期刊也是合适的(不是CNS级别的开创性工作,但是有清晰增量贡献的扎实工作)。
但它的天花板被以下几点限制:
- 缺少体内验证 → 限制了临床转化潜力
- 缺少功能性证明 → 限制了治疗靶点的说服力
- 机制研究只走到"转录因子层",没有深入"为什么β-catenin抑制了这些TF" → 限制了科学深度
- 完全没有讨论肝脏分区的生理背景 → 限制了生物学解读的深度
对古川研究室的整体定位来说:这篇代表了研究室从"找下游靶基因"转向"系统性研究通路-代谢-临床"的方向。研究室的核心优势仍然是对Wnt信号在消化器癌中的深耕,而本文是这条主线上的最新成果。
个人思考
读完这篇论文后,我对古川研究室的研究风格有几点认识:
务实而非追新:不是追逐最新的单细胞或空间组学技术,而是把传统的"转录因子-启动子-报告基因"研究做到极致,再加入RNA-seq+ChIP-seq+代谢组学组合,构建严密的证据链
临床转化意识:每篇论文都会引用TCGA数据做临床关联,说明研究室对临床意义高度重视——这与古川教授作为临床医生出身的背景一致
代际传承清晰:从2000年AXIN1的发现,到2017年HAL报告系统,再到2024年HAL机制揭示,可以看到研究室的工作是有"主线"的累积,而非散点式发表
可以作为博士论文的潜在切入点——本文留下了大量未解决的问题(CEBPA/FOXA1上游机制、体内验证、功能性证明、临床预后),任何一个都足够支撑一个完整的博士课题。如果我加入研究室,可以考虑围绕"Wnt信号抑制CEBPA/FOXA1的具体分子机制"作为博士课题切入点
面试可能被问到的问题
准备回答
- 你认为这篇论文最大的贡献和最大的局限分别是什么?
- 如果让你设计后续实验,你会优先做哪一个?为什么?
- Wnt信号在大肠癌和肝癌中调控的下游基因不同(如HAL和ARG1只在肝癌中被抑制),你认为这种"组织特异性"的根本原因是什么?
- 你对体内验证缺失这一点怎么看?这是否影响你对论文结论的接受程度?
- 本文与2014年Yamada教授那篇"重编程致癌"论文都讨论了"表观/转录调控异常驱动癌症",你认为两者的方法论有什么共通之处?