Appearance
体内重编程驱动Kras诱导的癌症发展(2018)
论文信息
教授: Yasuhiro Yamada · 大学: 东京大学 · 阅读日期: 2026-04-05
基本信息
| 项目 | 内容 |
|---|---|
| 标题 | In vivo reprogramming drives Kras-induced cancer development |
| 作者 | Hirofumi Shibata, Shingo Komura et al. |
| 发表于 | Nature Communications |
| DOI/链接 | 原文 |
| 被引次数 | 76 |
总结概要
2014年Cell论文证明了"纯表观遗传异常可以独立致癌"。本篇论文更进一步,探究表观遗传改变如何与经典癌基因(Kras突变)协同驱动成人癌症——以胰腺导管腺癌(PDAC)为模型。核心发现:仅1-3天的短暂重编程因子表达就足以抑制胰腺腺泡细胞的增强子活性,导致细胞身份丢失;在Kras突变背景下,这种短暂的表观遗传扰动足以激活ERK信号通路并快速诱发PDAC。维持腺泡细胞身份(强制表达Ptf1a或Mist1)可以阻止癌变。
研究背景与动机
- 2014年Cell论文证明了表观遗传异常可以独立驱动儿童癌症(Wilms瘤样肿瘤)。但成人癌症通常携带明确的驱动基因突变(如Kras),表观遗传改变在成人癌症中扮演什么角色?
- 胰腺癌(PDAC)是成人最致命的癌症之一(5年生存率仅3-5%)。经典模型认为PDAC通过基因突变的逐步累积发展:KRAS突变→CDKN2A丢失→TP53失活→SMAD4失活。但一个关键矛盾是:Kras突变在正常人的胰腺中也能被检测到,而且基因工程小鼠中Kras突变的胰腺细胞大部分在数周甚至数月内都保持正常——说明Kras突变本身不足以致癌,还需要"额外事件"
- 这篇论文的核心问题:重编程相关的表观遗传改变是否就是那个"额外事件"?它能否与Kras突变协同,快速启动胰腺癌?
核心方法
整体思路
与第一篇论文类似的"制造模型→观察结果→深挖机制"思路,但增加了两个重要维度:
- 引入经典癌基因突变(Kras^G12D、p53^R172H)与重编程的组合
- 新增ChIP-seq技术来分析增强子(enhancer)层面的表观遗传变化
- 增加"反向验证"——通过强制维持细胞身份来阻止癌变
制作模型
本篇论文构建了多个小鼠模型,复杂度远超第一篇。核心技术仍然是Tet-on系统+Cre-LoxP系统的组合。
新增的Cre-LoxP系统:
Cre-LoxP是另一种广泛使用的条件性基因操控系统,与Tet-on互补:
基因组中: [LoxP]---[Stop序列]---[LoxP]---[目标基因(如KrasG12D)]
无Cre时: Stop序列阻断转录 → 目标基因不表达(沉默)
有Cre时: Cre重组酶识别两个LoxP位点 → 切除中间的Stop序列 → 目标基因永久表达关键区别:Tet-on是可逆的(加Dox开启,撤Dox关闭),而Cre-LoxP是不可逆的(一旦Cre切除Stop,目标基因永久打开)。
在这篇论文中的组合使用:
Cre-LoxP → 永久激活Kras突变和p53突变(模拟人类癌症的基因背景)
Tet-on → 可逆控制重编程因子OSKM的表达(模拟表观遗传扰动)Pdx1-ires-Cre: Pdx1是胰腺特异性转录因子,将Cre重组酶连接在Pdx1后面,确保Cre只在胰腺细胞中表达 → 实现胰腺特异性的基因操控。
论文中的主要小鼠模型:
| 模型名称 | 携带的遗传改造 | 用途 |
|---|---|---|
| KC | Kras^G12D + Pdx1-Cre | 仅Kras突变的对照 |
| KPC | Kras^G12D + p53^R172H + Pdx1-Cre | Kras+p53双突变的对照 |
| C-OSKM | Pdx1-Cre + Rosa LSL-rtTA3 + Col1a1::tetO-OSKM | 无Kras突变 + 可控OSKM表达 |
| KC-OSKM | 同C-OSKM + Kras^G12D | Kras突变 + 可控OSKM表达(核心模型) |
| KPC-OSKM | 同KC-OSKM + p53^R172H | Kras+p53双突变 + 可控OSKM表达 |
C-OSKM模型的遗传构造详解(Fig 2a)
这个模型需要理解三个遗传元件如何协同工作:
元件1:Pdx1-ires-Cre
- Pdx1在胰腺细胞中天然表达 → Cre只在胰腺细胞中产生
元件2:Rosa26 LSL-rtTA3
- Rosa26位点插入了 [LoxP]-[Stop]-[LoxP]-[rtTA3]
- 只有当Cre切除Stop后,rtTA3才能表达
- 因为Cre只在胰腺中有 → rtTA3只在胰腺中表达
元件3:Col1a1::tetO-OSKM-ires-mCherry
- 和第一篇论文一样的TetOP-OSKM盒
- 但现在rtTA3只在胰腺中有 → OSKM只能在胰腺中被Dox诱导
最终效果: 给Dox → 只有胰腺细胞表达OSKM(全身其他组织不受影响) 这比第一篇论文的全身性OSKM表达更精确,排除了其他组织的干扰。
实验流程
4周龄小鼠 → 给Dox 3天(短暂开启胰腺中的OSKM)→ 撤Dox 7天 → Day 10观察结果与第一篇论文的关键区别:
- 第一篇:全身OSKM表达,7天Dox → Wilms瘤样肾脏肿瘤
- 本篇:仅胰腺OSKM表达,仅3天Dox → 在Kras突变背景下快速发展为PDAC
实验与结果
第一步:证明Kras/p53突变本身不足以致癌(Fig 1)
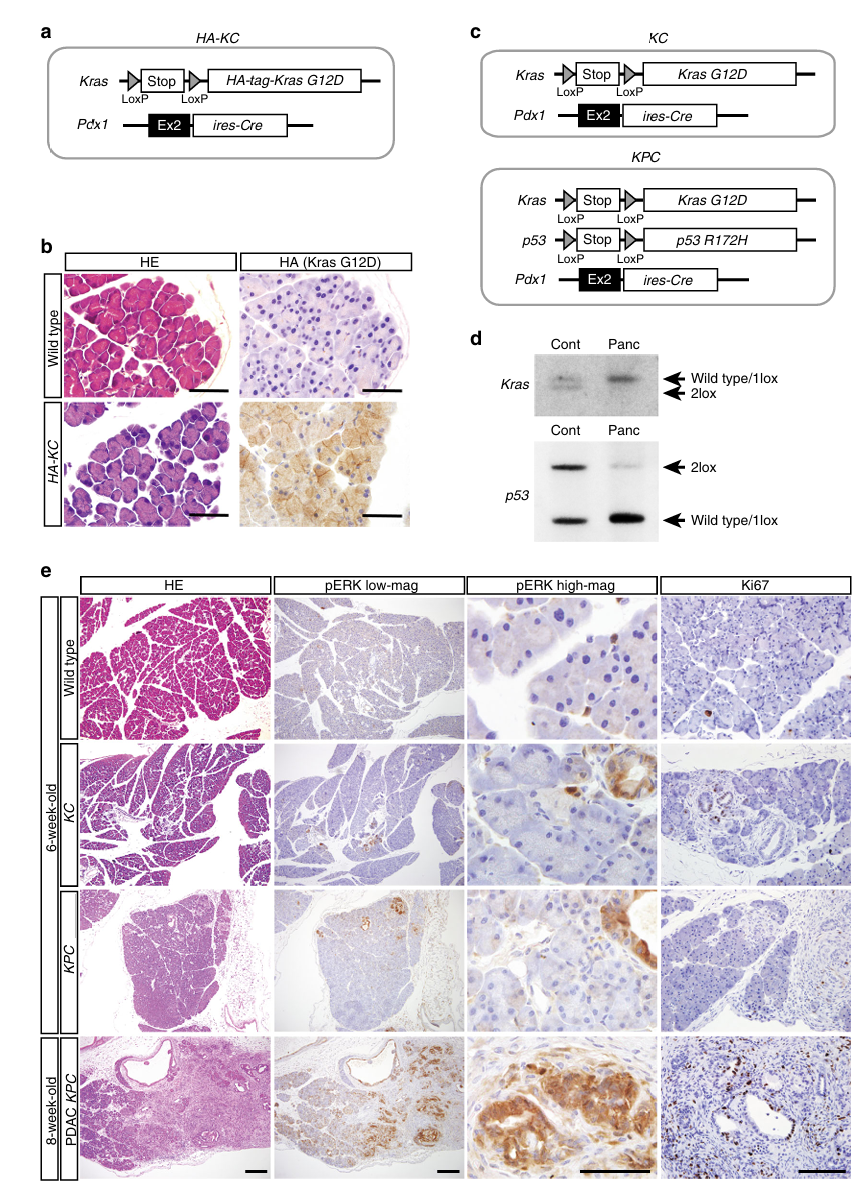
方法:H&E + pERK免疫组化 + Ki67免疫组化
pERK是什么? ERK(细胞外信号调节激酶)是Ras-MAPK信号通路的核心效应分子。Kras突变理论上应该持续激活这条通路。pERK(磷酸化ERK)= ERK被激活的标志。
核心发现:
KC小鼠(仅Kras突变)6周龄:
- H&E:大部分胰腺组织正常,仅有零星早期PanIN
- pERK:绝大部分胰腺细胞pERK阴性 → ERK通路没有被激活!
- Ki67:绝大部分胰腺细胞Ki67阴性 → 没有异常增殖
KPC小鼠(Kras + p53双突变)6周龄:
- 同样,大部分胰腺细胞正常
- pERK仅在少数PanIN/PDAC病灶中阳性
- 8周龄出现局灶性PDAC,但范围有限这个发现的重要性: Kras^G12D突变蛋白确实在胰腺细胞膜上表达(HA标签免疫染色证实),但下游ERK通路却没有被激活。即使加上p53突变也不够。这直接说明:基因突变不等于信号激活,还需要额外的事件来"解锁"突变的致癌潜力。
第二步:短暂重编程导致可逆的腺泡-导管化生(Fig 2)
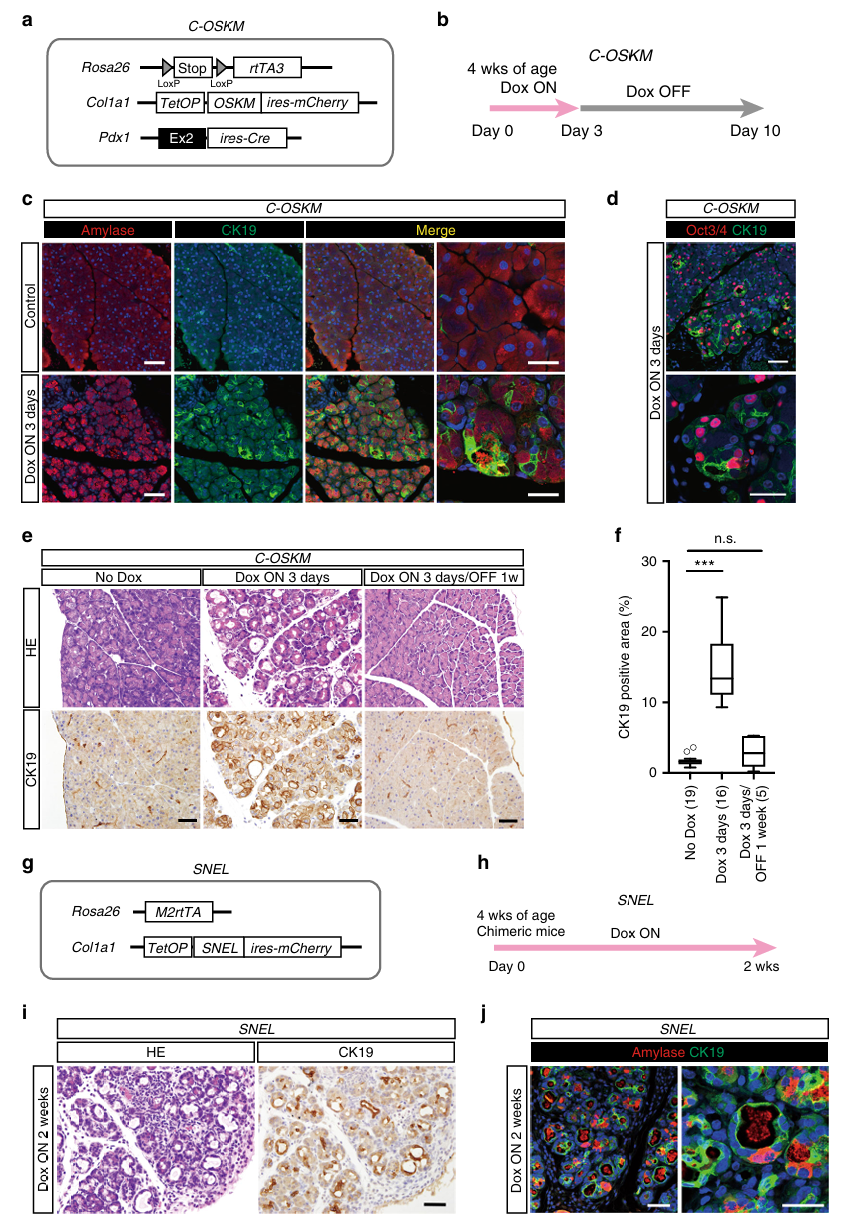
ADM是什么?
腺泡-导管化生(Acinar to Ductal Metaplasia, ADM):胰腺腺泡细胞(分泌消化酶的细胞)转变为导管样细胞的过程。这被认为是胰腺癌发展的早期事件。
在人类中,慢性胰腺炎中就可以观察到ADM,而胰腺炎是PDAC的已知危险因素。
方法:H&E + Amylase/CK19双重免疫荧光
- Amylase(淀粉酶):腺泡细胞标记(腺泡细胞分泌消化酶)
- CK19(细胞角蛋白19):导管细胞标记
核心发现:
C-OSKM小鼠,Dox 3天:
- H&E:腺泡腺体扩张
- CK19在腺泡区域出现阳性(正常情况下腺泡区不该有CK19)
- Amylase和CK19在同一细胞中共表达 → 腺泡细胞正在转变为导管样细胞(ADM)
- Oct3/4在CK19阳性细胞中表达 → 证实是重编程因子驱动了ADM
C-OSKM小鼠,Dox 3天 → 撤Dox 1周:
- H&E:胰腺组织基本恢复正常
- CK19阳性区域几乎消失
→ ADM是**可逆的**(因为腺泡细胞保留了表观遗传记忆)SNEL对照实验(Fig 2g-j): 用另一套不含Klf4的重编程因子(Sall4, Nanog, Esrrb, Lin28 = SNEL)也能诱导ADM → 证明ADM不是某个特定因子(如Klf4)的个别效应,而是重编程过程本身导致的。
第三步:重编程因子抑制了腺泡细胞增强子(Fig 3)——机制层面
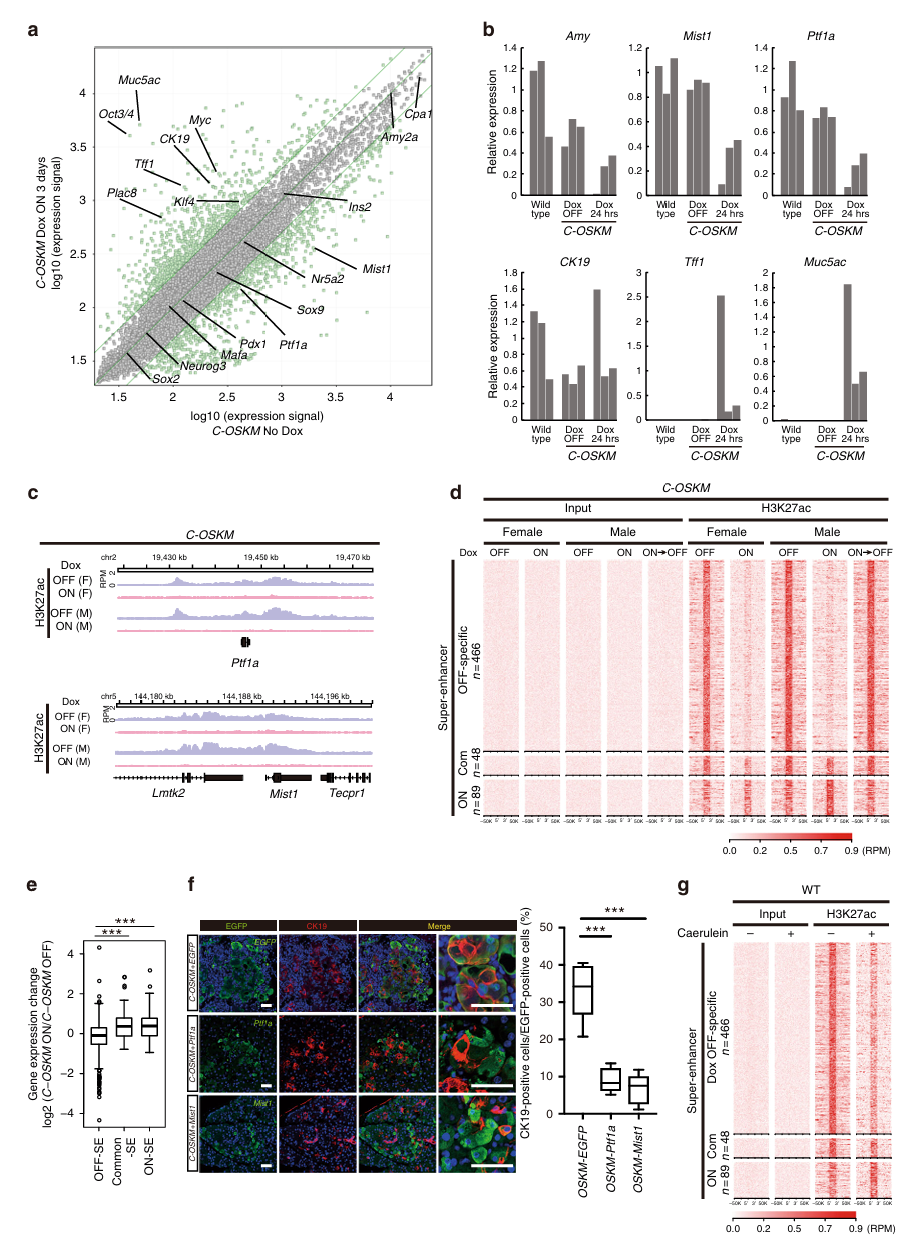
新方法:ChIP-seq
ChIP-seq是什么?
ChIP = 染色质免疫沉淀(Chromatin Immunoprecipitation)。简单来说,就是用抗体去"钓"与特定蛋白(或特定组蛋白修饰)结合的DNA片段,然后测序看这些DNA片段来自基因组的哪些位置。
活体组织 → 甲醛固定(把蛋白和DNA"冻结"在一起)
↓
超声打碎DNA为小片段
↓
加入针对H3K27ac的抗体 → 抗体"钓"出带有H3K27ac修饰的DNA片段
↓
去除蛋白,纯化DNA
↓
高通量测序 → 得到这些DNA片段在基因组上的位置
↓
在基因组浏览器上看到"峰"(peak)= H3K27ac富集的位置 = 活跃增强子H3K27ac是什么? 组蛋白H3的第27位赖氨酸被乙酰化。这是活跃增强子(active enhancer)的标记。增强子是基因组中远离启动子但可以增强基因表达的调控元件。一个基因的增强子上H3K27ac信号强 → 该增强子活跃 → 对应基因倾向于高表达。
超级增强子(Super Enhancer, SE): 一种特别强的增强子区域,由多个增强子聚集而成,H3K27ac信号特别高。超级增强子通常控制着决定细胞身份的关键基因。
核心发现:
Fig 3a-b(微阵列 + qRT-PCR):
C-OSKM小鼠 Dox 3天:
- 腺泡细胞关键转录因子 Ptf1a、Mist1 → 显著下调
- 导管标记 CK19 和 ADM相关基因 Muc5ac、Tff1 → 上调
C-OSKM小鼠 Dox仅24小时:
- Ptf1a、Mist1已经开始明显下调
- CK19和ADM基因已经开始上调
→ 腺泡细胞身份的丢失发生得非常快(24小时内)Fig 3c-d(ChIP-seq,最关键的机制数据):
Ptf1a基因的增强子区域:
Dox OFF → H3K27ac信号强(增强子活跃 → Ptf1a正常表达)
Dox ON 3天 → H3K27ac信号显著降低(增强子被抑制 → Ptf1a沉默)
Mist1基因的增强子区域:同样变化
全基因组层面:
Dox OFF特异性超级增强子(466个)→ Dox ON后信号大幅下降
→ 腺泡细胞的超级增强子被全面抑制
→ 撤Dox后H3K27ac恢复(可逆!与ADM可逆一致)Fig 3e(超级增强子与基因表达的关联):
- 与Dox OFF特异性SE关联的基因:Dox处理后表达显著下降
- 与共有SE或Dox ON特异性SE关联的基因:不受影响 → OSKM选择性地抑制了腺泡细胞身份相关的超级增强子
Fig 3f(反向验证——强制维持腺泡身份可以阻止ADM):
- 在表达OSKM的同时,强制表达Ptf1a或Mist1
- 结果:CK19阳性(ADM)细胞比例显著降低 → 证实了腺泡增强子的抑制是ADM发生的直接原因
Fig 3g(与临床的连接——胰腺炎模型):
- 用caerulein(一种CCK类似物)诱导胰腺炎
- ChIP-seq发现:caerulein处理同样导致腺泡细胞超级增强子的抑制,模式与OSKM诱导相似 → 炎症刺激通过类似重编程的增强子抑制机制导致ADM → 这解释了为什么胰腺炎是PDAC的危险因素
第四步:重编程 + Kras突变 → 快速PDAC发展(Fig 4)——核心发现
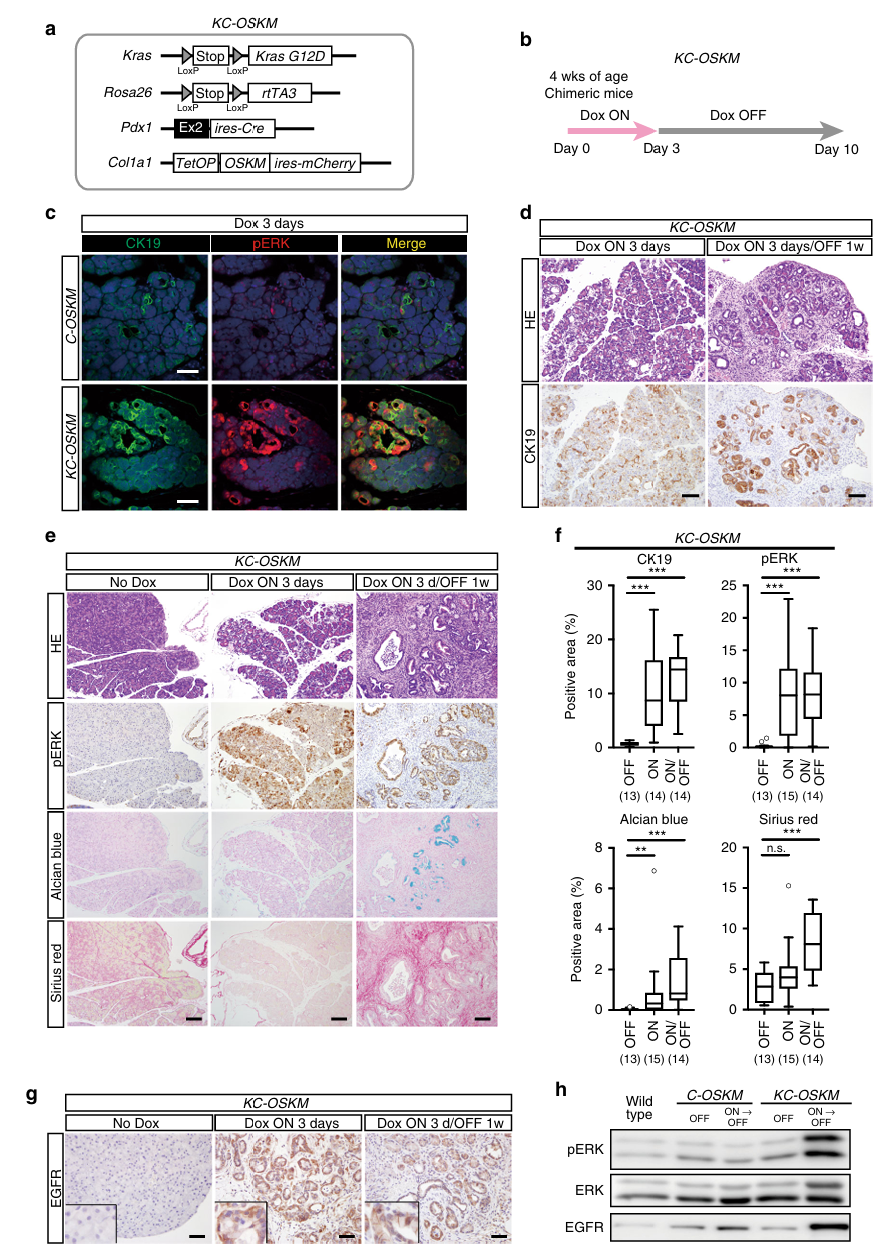
方法:CK19/pERK双重免疫荧光 + H&E + Alcian blue + Sirius red + EGFR免疫组化 + Western blot
新的染色方法说明:
- Alcian blue(阿利新蓝):PanIN标记(PanIN细胞分泌黏液,Alcian blue染黏液)
- Sirius red(天狼星红):纤维化标记(胰腺癌特征性的间质纤维化/结缔组织增生)
- EGFR:表皮生长因子受体,Ras-MAPK通路的上游激活信号
核心发现:
Fig 4c(C-OSKM vs KC-OSKM,Dox 3天):
C-OSKM(无Kras突变)Dox 3天:
- CK19阳性(ADM发生)
- pERK:仅微弱阳性 → ERK通路轻度激活
KC-OSKM(有Kras突变)Dox 3天:
- CK19阳性(ADM同样发生)
- pERK:强阳性!→ ERK通路被强烈激活
→ Kras突变 + 表观遗传扰动 = ERK通路的协同强激活Fig 4d-f(KC-OSKM,Dox 3天→撤Dox 1周):
这是全文最关键的对比:
C-OSKM撤Dox 1周后:
- CK19恢复阴性 → ADM逆转,胰腺恢复正常 ✓
KC-OSKM撤Dox 1周后:
- CK19持续阳性 → ADM不可逆!✗
- pERK持续强阳性 → ERK通路持续激活
- Alcian blue阳性 → PanIN形成
- Sirius red阳性 → 大面积纤维化
- 组织学:PanIN/PDAC
→ **短短10天内(3天Dox + 7天撤Dox),从正常胰腺变成了PDAC!**Fig 4g-h(EGFR正反馈机制):
KC-OSKM Dox 3天 → EGFR表达上调
KC-OSKM 撤Dox后 → EGFR表达持续升高(不可逆)
C-OSKM 撤Dox后 → EGFR恢复正常
Western blot确认:KC-OSKM撤Dox后,pERK和EGFR持续高表达机制解读: EGFR是ERK通路的上游激活因子。在Kras突变背景下,ADM诱导的ERK轻度激活→促进EGFR表达→EGFR进一步激活ERK→形成正反馈环路→ERK通路持续激活→不可逆的癌变。没有Kras突变时,这个正反馈不够强,撤Dox后系统恢复正常。
第五步:p53突变加速PDAC发展(Fig 5a-e)
KPC-OSKM(Kras + p53 + OSKM)vs KC-OSKM(Kras + OSKM):
KC-OSKM Dox 3天/撤Dox 1周:混合病变(PanIN + PDAC)
KPC-OSKM Dox 3天/撤Dox 1周:几乎全是PDAC(更恶性)
KPC-OSKM小鼠在3个月内死亡,部分出现癌性腹水
从肿瘤建立的细胞系可以在裸鼠皮下形成继发肿瘤→ p53突变促进了从PanIN到PDAC的进展(与人类胰腺癌多步进展模型一致)
第六步:维持腺泡细胞身份可以阻止Kras驱动的癌症(Fig 5f-h)——反向验证
实验设计:
- 在KC小鼠(Kras突变)中,用caerulein诱导胰腺炎(模拟临床情况)
- 同时用Dox诱导Ptf1a或Mist1(强制维持腺泡细胞身份)
- 对照组:Dox诱导mCherry(无功能对照)
结果:
KC-mCherry + caerulein → PanIN形成 + pERK强阳性(癌前病变)
KC-Ptf1a + caerulein → PanIN几乎不形成 + pERK显著降低
KC-Mist1 + caerulein → 同上→ 强制维持腺泡细胞的增强子活性(通过表达腺泡特异性转录因子),可以阻止Kras突变细胞的癌变 → 这提供了一个全新的抗癌思路:不是去直接杀死癌细胞,而是维持细胞的分化身份来防止癌变
第七步:延长重编程→不同类型的癌症(Fig 6)
KC-OSKM Dox 3天 → PDAC(早期重编程 = 腺泡增强子抑制 → 分化身份丢失)
KC-OSKM Dox 1-2周 → 未分化癌,表达Sall4、Lin28a(晚期重编程 = 部分获得ESC特征)
→ 在胃部也发展出未分化癌
→ 类似人类AFP产生性胃癌(一种极恶性的成人癌症)验证:人类AFP产生性胃癌样本中,SALL4、LIN28A、LIN28B频繁共表达,ESC-Core和Myc模块部分激活 → 与小鼠模型表型一致
→ 重编程的不同阶段(早期 vs 晚期)对应不同类型的癌症
关键发现
- Kras/p53突变本身不足以激活ERK通路和诱发PDAC → 需要额外的表观遗传事件
- 仅1-3天的OSKM表达足以抑制腺泡细胞超级增强子 → 导致细胞身份丢失(ADM)
- 表观遗传扰动 + Kras突变 = ERK通路的持续激活 + 10天内快速PDAC发展
- 炎症(caerulein/胰腺炎)通过类似机制抑制腺泡增强子 → 解释了胰腺炎作为PDAC危险因素的分子基础
- 维持腺泡细胞身份(Ptf1a/Mist1表达)可以阻止Kras驱动的癌变 → 新的抗癌思路
- 重编程的不同阶段产生不同类型的癌症 → 早期重编程→PDAC;晚期重编程→AFP产生性未分化癌
证据链
① Kras突变 + 正常表观遗传 → 大部分细胞正常(不致癌)
② 重编程 + 无Kras突变 → 可逆的ADM(不致癌)
③ 重编程 + Kras突变 → 不可逆ADM + ERK持续激活 + 快速PDAC(致癌!)
④ 炎症也能抑制腺泡增强子 → 与重编程效果一致 → 临床相关性
⑤ 强制维持细胞身份 → 阻止癌变 → 反向验证因果关系
结论:表观遗传改变(腺泡增强子的丢失)是Kras驱动型胰腺癌启动的关键"解锁"事件与研究室其他工作的关系
这篇论文是2014年Cell论文的直接延伸,构成了逻辑链的第二环:
Cell 2014 → 表观遗传异常可以独立致癌(纯表观遗传驱动的儿童癌症模型)
↓
Nat Commun 2018(本篇)→ 表观遗传改变与经典癌基因协同驱动成人癌症
↓
Cell Reports 2022 → 反向利用重编程技术发现癌症治疗靶点(转化应用)本篇的独特贡献在于:将"表观遗传驱动癌症"的概念从一个相对特殊的场景(纯表观遗传→儿童癌症)拓展到了临床上最常见、最致命的成人癌症(Kras突变型胰腺癌),大大提升了这一研究方向的临床意义和影响力。
面试可能被问到的问题
准备回答
- 为什么Kras突变本身不能激活ERK通路?腺泡细胞的什么特性在"压制"Kras的致癌信号?
- 重编程因子抑制增强子和炎症抑制增强子的机制是否完全一样?还是只是最终结果相似?
- 临床上,能否通过检测腺泡增强子活性来预测胰腺癌风险?
- 维持细胞身份作为抗癌策略,在临床上可行吗?有什么局限性?
- 这篇论文和2014年Cell论文的核心区别是什么?各自的贡献和局限性?
个人思考
相比起之前2014年的文章,本文更加深入探究了重编程与传统致癌途径的关联性。也间接揭示了一种新的抗癌思路。
但是我感觉这篇文章的证据链没有上一篇那么明确,因为条件的控制比较有限。