Appearance
癌基因依赖的重编程抵抗性揭示癌症治疗靶点(2022)
论文信息
教授: Yasuhiro Yamada · 大学: 东京大学 · 阅读日期: 2026-04-10
基本信息
| 项目 | 内容 |
|---|---|
| 标题 | The oncogene-dependent resistance to reprogramming unveils cancer therapeutic targets |
| 作者 | Kenji Ito, Kohei Nagata, Sho Ohta et al. |
| 发表于 | Cell Reports |
| DOI/链接 | 原文 |
| 被引次数 | - |
总结概要
前两篇论文证明了"重编程可以导致癌症",这篇论文则反过来利用重编程技术来发现癌症的治疗靶点。核心发现:癌细胞之所以对重编程高度抵抗,是因为驱动癌基因将重编程因子"劫持"到了癌症特异性增强子上,阻断了向多能性方向的转录应答。利用这种"癌基因依赖的重编程抵抗性",研究者建立了一种新型药物筛选平台——通过检测哪种药物能解除重编程抵抗(恢复NANOG表达),来发现该癌症的驱动信号通路。通过这一策略,成功发现mTOR信号通路是透明细胞肉瘤(CCS)的治疗靶点。
研究背景与动机
- 前两篇论文探讨的都是"重编程→癌症"的方向。但反过来思考:为什么癌细胞对重编程如此抵抗? 这种抵抗性的分子机制是什么?能不能利用这种抵抗性来做有用的事情?
- 众所周知,癌细胞比正常体细胞更难被重编程为iPSC。虽然有少数成功案例(如CML、黑色素瘤),但效率极低。此前无人系统研究过癌细胞抵抗重编程的机制
- **透明细胞肉瘤(Clear Cell Sarcoma, CCS)**是一种罕见但致命的软组织肉瘤,由EWS/ATF1融合基因驱动。目前没有有效的靶向治疗药物。此前山田研究室已证明CCS细胞可以被重编程为iPSC(Komura et al., 2019 Nat Commun),但需要关闭EWS/ATF1表达
- 核心问题:癌基因是如何阻止重编程的?能否利用"解除重编程抵抗"作为筛选指标,找到癌症的驱动信号通路和治疗靶点?
核心方法
整体思路
与前两篇论文完全不同的研究思路——从体内小鼠模型转向体外细胞系筛选:
前两篇的逻辑:重编程 → 表观遗传改变 → 导致癌症(重编程是"因")
本篇的逻辑: 癌细胞 → 抵抗重编程 → 利用这种抵抗性来筛选药物(重编程是"工具")具体分为四步:
- 发现现象:癌基因阻止了癌细胞的重编程
- 揭示机制:癌基因将重编程因子"劫持"到癌症特异性增强子
- 建立筛选平台:用NANOG表达作为"重编程解除"的指标
- 应用:发现CCS的治疗靶点(mTOR通路)
关键实验系统
G1297 CCS细胞系: 这是山田研究室此前建立的小鼠CCS细胞系,具有一个关键特性——EWS/ATF1的表达可以被Dox控制(Dox-controllable)。
加Dox → EWS/ATF1表达 → CCS状态(癌细胞)
撤Dox → EWS/ATF1关闭 → 失去CCS特征这让研究者可以在同一种细胞中,通过简单的开/关EWS/ATF1,对比有/无癌基因时重编程的反应差异。
OSKM可诱导的人类癌细胞系: 用piggyBac转座子系统将Dox诱导的OSKM盒(小鼠OSKM)整合到人类癌细胞基因组中。
加Dox → OSKM表达 → 观察重编程早期转录应答(主要看NANOG表达)构建了多种OSKM可诱导的人类癌细胞系:HCC827(EGFR突变肺癌)、SK-BR3(HER2扩增乳腺癌)、K562(BCR-ABL融合CML)、A549(KRAS突变肺癌)、MP-CCS-SY和KAS(人CCS细胞系)、LCC-028(EML4-ALK融合肺癌患者来源细胞PDC)等。
新方法:高通量药物筛选与多重qRT-PCR
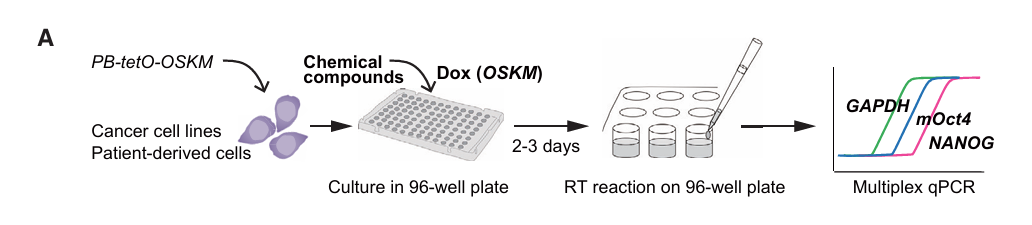
筛选系统设计(Fig 4A):
96孔板中培养OSKM可诱导的癌细胞
↓ 同时加入Dox(开启OSKM)+ 待筛选化合物
↓ 培养2-3天
↓ 直接在96孔板中裂解细胞、逆转录
↓ 用TaqMan探针同时检测三个基因(多重qPCR):
- NANOG(重编程早期应答指标 → 如果上升,说明重编程抵抗被解除)
- mOct4(外源OSKM转基因表达水平 → 排除化合物影响OSKM表达的情况)
- GAPDH(内参基因 → 归一化)
↓
筛选标准:NANOG/GAPDH > 2 且 NANOG/mOct4 > 2 的化合物
→ 候选驱动信号通路抑制剂为什么用NANOG而不是直接看细胞是否死亡?
这是本文最巧妙的设计。传统药物筛选看的是"哪种药能杀死癌细胞"(看增殖/存活)。但这种方法会筛出很多非特异性的细胞毒药物。而NANOG表达的上升反映的是癌细胞特异性转录网络被扰动——只有抑制了真正的驱动信号通路,才能解除癌细胞的"转录僵硬性",让重编程因子发挥作用。
论文Fig 4F直接证明了这一点:大多数抑制细胞增殖的化合物并不诱导NANOG表达,说明NANOG筛选比传统增殖筛选更能富集到驱动信号通路的抑制剂。
实验与结果
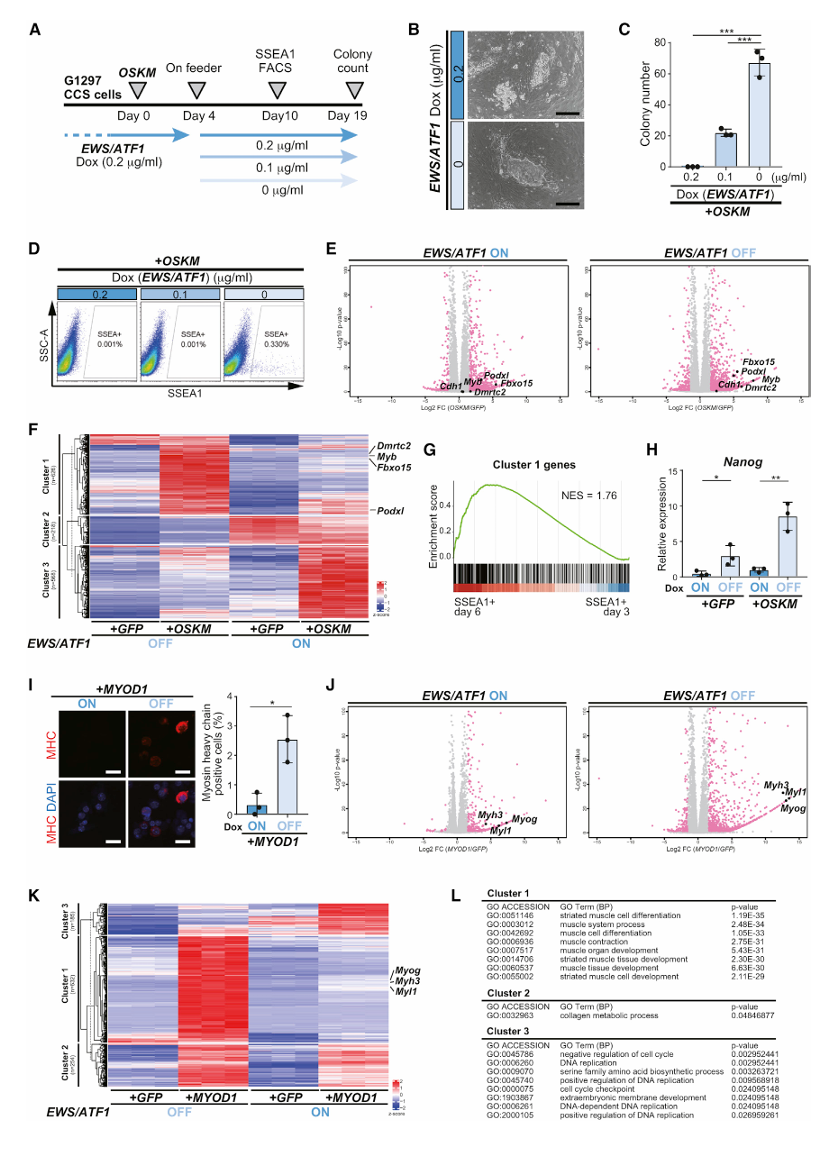
第一步:EWS/ATF1阻止了CCS细胞的重编程(Fig 1A-H)
方法:逆转录病毒转导OSKM + iPSC集落计数 + FACS(SSEA1)+ RNA-seq + qRT-PCR
核心发现:
G1297 CCS细胞 + OSKM:
EWS/ATF1 ON(加Dox 0.2μg/ml)→ 不形成iPSC样集落
EWS/ATF1 OFF(不加Dox)→ 形成iPSC样集落
EWS/ATF1表达量与iPSC集落数量呈反比关系(Fig 1C)
SSEA1+细胞(重编程早期标记):
EWS/ATF1 ON → SSEA1+细胞极少(重编程早期就被阻断)
EWS/ATF1 OFF → SSEA1+细胞明显增加→ EWS/ATF1阻断了重编程的早期阶段
RNA-seq揭示转录应答的差异(Fig 1E-F):
EWS/ATF1 OFF + OSKM → 一组基因被上调(Cluster 1),包含重编程早期标记
如Fbxo15、Podxl、Myb、Dmrtc2
EWS/ATF1 ON + OSKM → 这些基因的上调程度大打折扣
同时有另一组基因被异常上调(Cluster 3)→ 癌基因不仅抑制了正常的重编程转录应答,还诱导了异常的转录应答
MYOD1转分化实验(Fig 1I-L)——重要对照:
MYOD1是肌肉分化的主调控因子,可以将成纤维细胞直接转变为肌肉细胞。研究者发现EWS/ATF1同样阻止了MYOD1介导的肌肉转分化:
- EWS/ATF1 ON → MHC+(肌球蛋白重链阳性)细胞很少
- EWS/ATF1 OFF → MHC+细胞显著增加
- 肌肉分化相关基因(Myog、Myh3等)在EWS/ATF1 OFF时上调更明显
→ 癌基因阻止的不只是向多能性方向的重编程,而是广泛地阻止了细胞命运的改变 → 癌基因维持了癌细胞身份的"僵硬性"
第二步:癌基因将重编程因子"劫持"到癌症特异性增强子(Fig 2)——机制层面
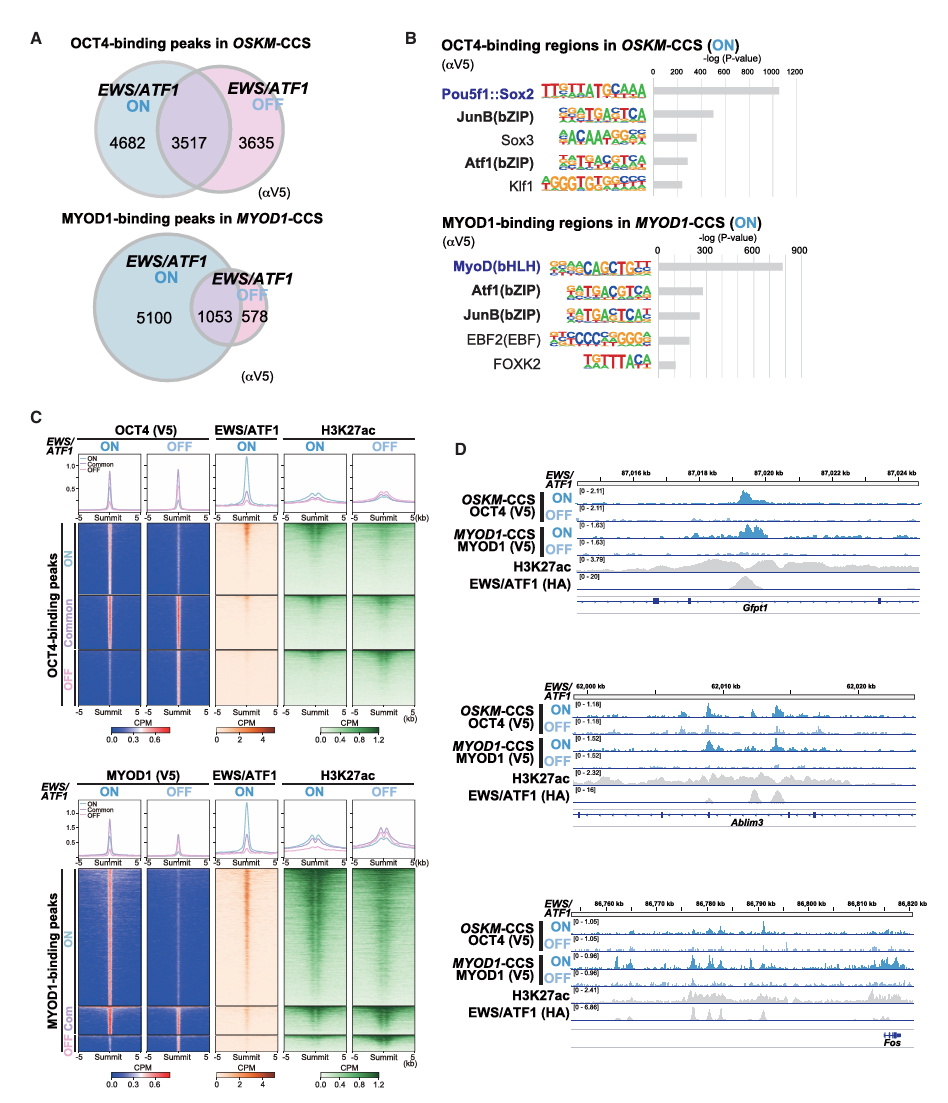
方法:ChIP-seq(V5标签标记的外源OCT4和MYOD1)+ 基序分析(Motif analysis)
核心发现——ChIP-seq(Fig 2A):
一个出乎意料的结果:
EWS/ATF1 ON时,OCT4在基因组上的结合位点数量反而更多(8199 vs 7152)
MYOD1同样如此(6153 vs 1631)→ 癌基因没有阻止重编程因子结合DNA,反而让它们结合到了更多的位置。问题不在于"结合不了",而在于**"结合到了错误的地方"**。
基序分析(Fig 2B)——揭示"劫持"机制:
EWS/ATF1 ON时,OCT4结合位点中:
- 不仅富集了OCT4自己的结合基序(Pou5f1::Sox2)
- 还富集了ATF/CRE相关基序 ← 这是EWS/ATF1的结合基序!
MYOD1结合位点同样:
- 富集了MyoD(bHLH)基序
- 也富集了Atf1(bZIP)和JunB(bZIP)基序→ 重编程因子被"拉"到了EWS/ATF1所在的位置
H3K27ac和EWS/ATF1的共定位(Fig 2C-D):
EWS/ATF1 ON时OCT4的独特结合峰:
- 与EWS/ATF1的结合位点高度重叠
- 这些位置H3K27ac信号强 → 是CCS的活跃增强子
EWS/ATF1 OFF时:
- OCT4不再结合到这些癌症增强子上
- OCT4转而结合到正常重编程过程应该结合的位点机制总结:
正常细胞的OSKM重编程:
OSKM → 结合体细胞增强子 → 抑制体细胞程序 → 激活多能性网络 → iPSC
癌细胞的OSKM重编程:
OSKM → 被癌基因"劫持"到癌症特异性增强子 → 无法有效抑制癌细胞程序
→ 多能性方向的转录应答受阻 → 重编程失败
关闭癌基因后:
OSKM → 不再被劫持 → 正常结合 → 重编程恢复这就像你想把车开到A地(多能性),但GPS(癌基因)一直把你导航到B地(癌症增强子),你永远到不了A。关掉错误的GPS,才能到达正确目的地。
第三步:其他癌症类型也存在同样的现象(Fig 3)——普适性验证
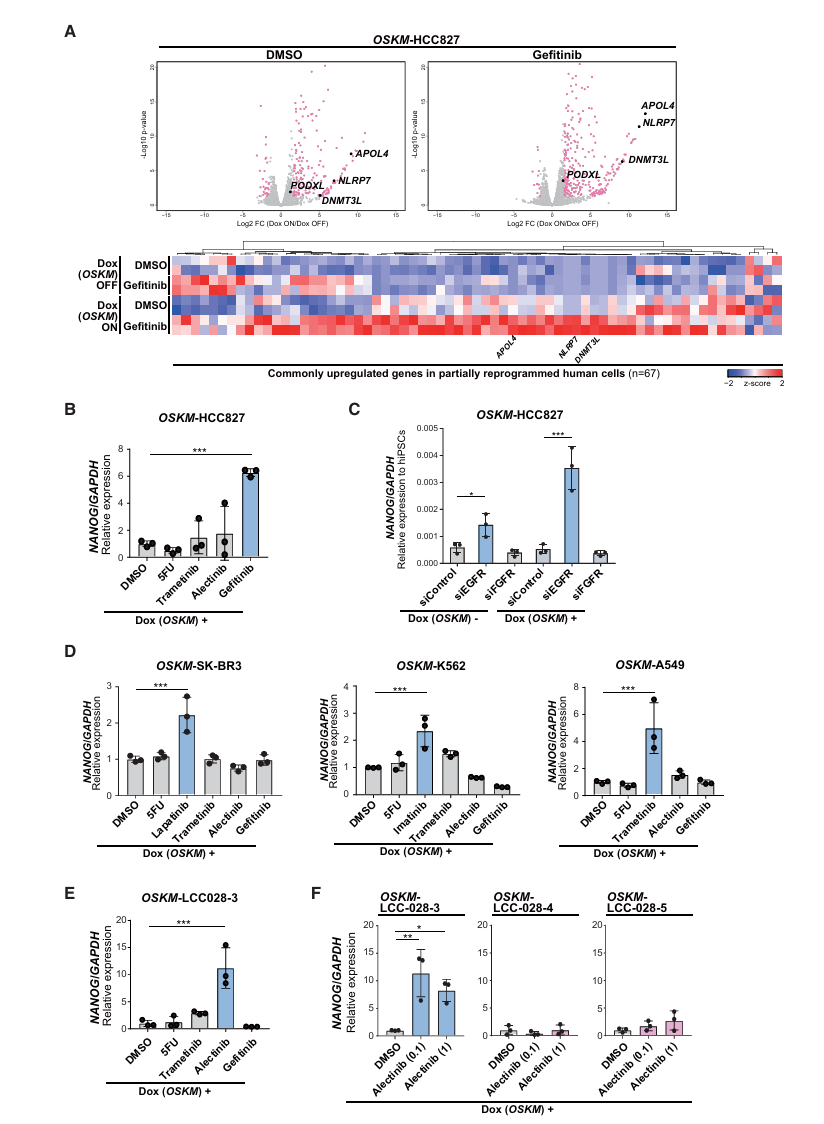
方法:构建OSKM可诱导的多种人类癌细胞系 + 用对应的靶向药处理 + qRT-PCR检测NANOG
核心发现:
| 癌细胞系 | 驱动癌基因 | 对应靶向药 | 加药后NANOG变化 |
|---|---|---|---|
| HCC827 | EGFR突变 | 吉非替尼(Gefitinib) | 显著上升 ✓ |
| SK-BR3 | HER2扩增 | 拉帕替尼(Lapatinib) | 显著上升 ✓ |
| K562 | BCR-ABL融合 | 伊马替尼(Imatinib) | 显著上升 ✓ |
| A549 | KRAS突变 | 曲美替尼(Trametinib) | 显著上升 ✓ |
关键对照:
- 用非对应的靶向药处理 → NANOG不变
- 用化疗药(5-FU)处理 → NANOG不变
- siRNA敲低EGFR → NANOG上升;敲低FGFR(非驱动基因)→ NANOG不变
→ 只有抑制真正的驱动癌基因信号,才能解除重编程抵抗 → 具有普适性
患者来源细胞(PDC)的验证(Fig 3E-F)——临床转化潜力:
LCC-028-3(EML4-ALK融合肺癌,对阿来替尼[Alectinib]临床敏感):
+ OSKM + Alectinib → NANOG显著上升 ✓
LCC-028-4、LCC-028-5(同一患者,获得性阿来替尼耐药后的细胞):
+ OSKM + Alectinib → NANOG不上升 ✗→ NANOG的响应与患者的临床药物敏感性完全一致! 耐药后重编程抵抗无法被同一药物解除 → 这意味着这个筛选系统可以用于个体化精准医疗
第四步:化合物库筛选——发现驱动信号通路(Fig 4-5)
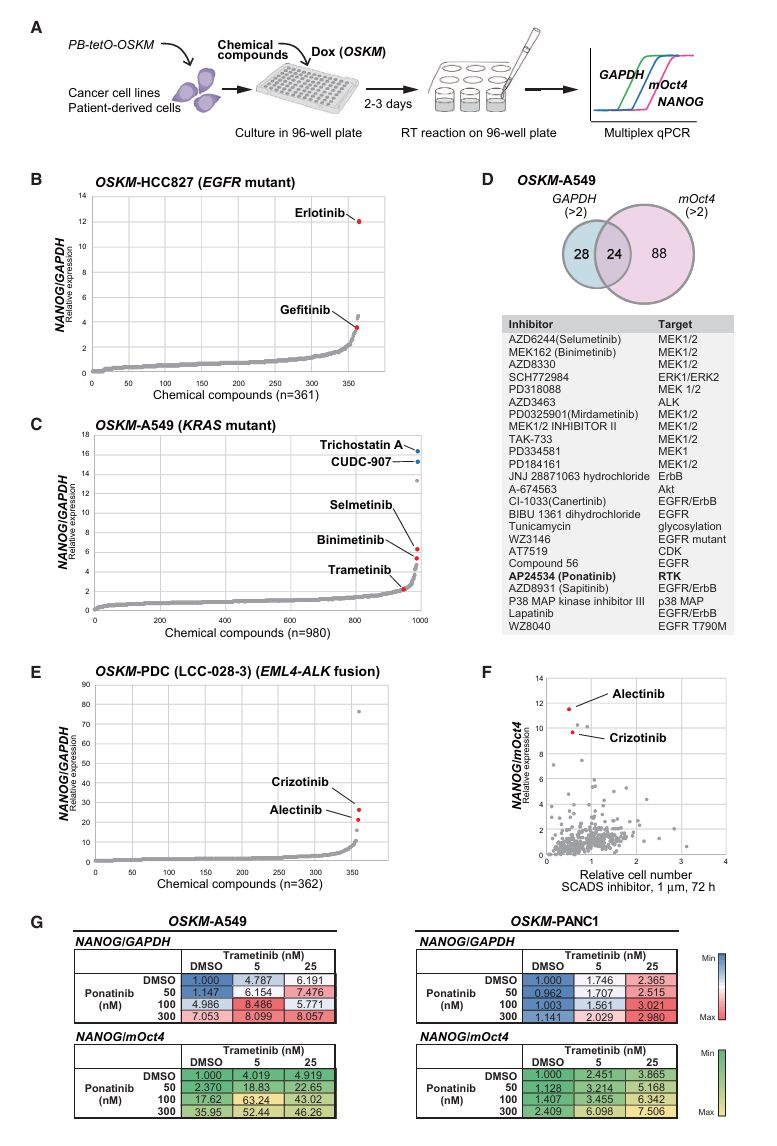
方法:SCADS抑制剂库(361-980种化合物)× OSKM可诱导的癌细胞系 → 96孔板多重qRT-PCR
验证性筛选(Fig 4B-F):
OSKM-HCC827(EGFR突变)筛选361化合物:
→ 筛出13个增强NANOG的化合物
→ 其中包含吉非替尼和厄洛替尼(两种EGFR抑制剂)✓
OSKM-A549(KRAS突变)筛选980化合物:
→ NANOG/GAPDH>2且NANOG/mOct4>2的24个化合物中
→ 绝大多数是KRAS相关信号通路抑制剂(MEK抑制剂为主)✓
OSKM-LCC-028-3(EML4-ALK融合)筛选361化合物:
→ 筛出12个化合物,包含ALK抑制剂克唑替尼和阿来替尼 ✓→ 筛选系统可以可靠地"钓"出已知的驱动信号通路抑制剂 → 系统验证成功
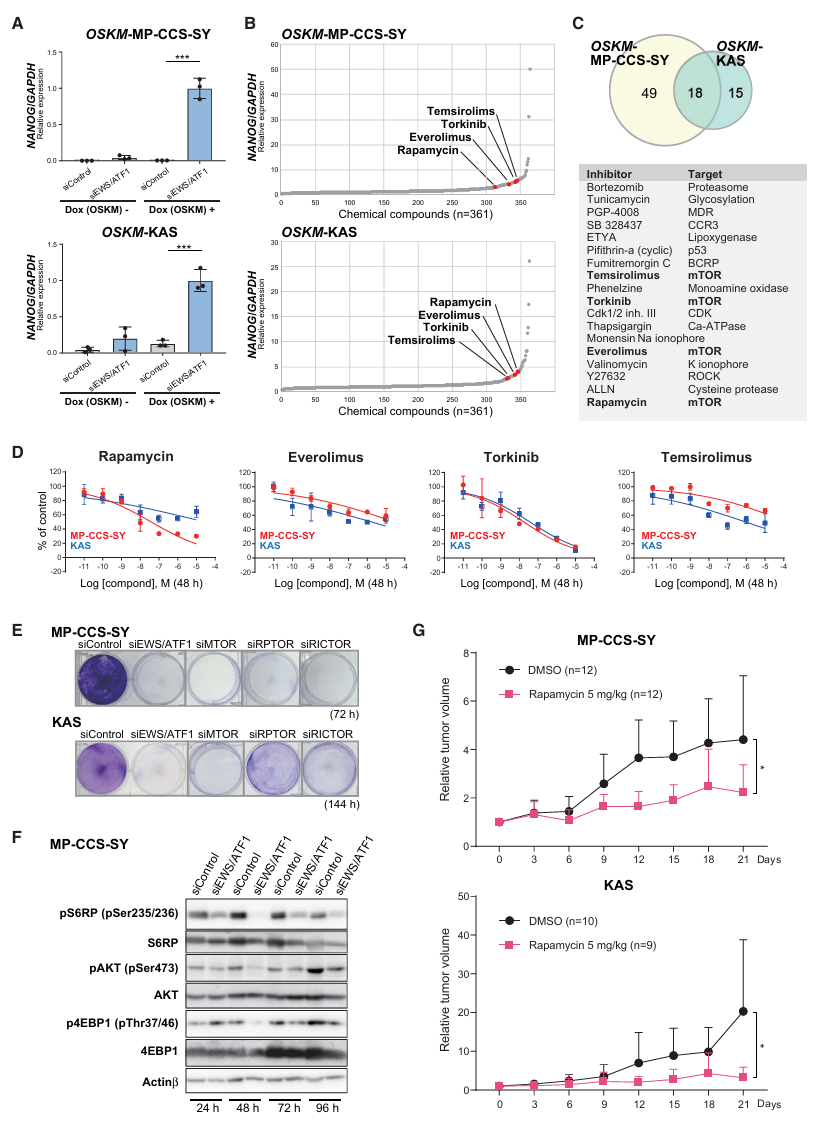
CCS的新靶点发现(Fig 5A-G)——核心转化成果:
CCS目前没有有效的靶向治疗,EWS/ATF1的下游效应通路不清楚。
OSKM-MP-CCS-SY和OSKM-KAS(两种人CCS细胞系)筛选361化合物:
→ 两个细胞系共同筛出18个增强NANOG的化合物
→ 其中所有4种mTOR抑制剂全部入选!
(Rapamycin、Everolimus、Torkinib、Temsirolimus)验证mTOR是EWS/ATF1的下游通路:
siRNA敲低EWS/ATF1 → mTOR下游靶点磷酸化降低:
- pS6RP ↓(mTORC1靶点)
- p4EBP1 ↓(mTORC1靶点)
- pAKT ↓(mTORC2靶点)
→ 证实EWS/ATF1确实激活了mTOR信号通路体内验证(Fig 5G):
CCS细胞异种移植到裸鼠:
DMSO对照组 → 肿瘤持续生长
Rapamycin 5mg/kg → 肿瘤生长显著抑制(MP-CCS-SY和KAS两种细胞系均有效)→ mTOR抑制剂是CCS的潜在治疗药物
第五步:联合用药筛选(Fig 6)——进一步优化治疗方案
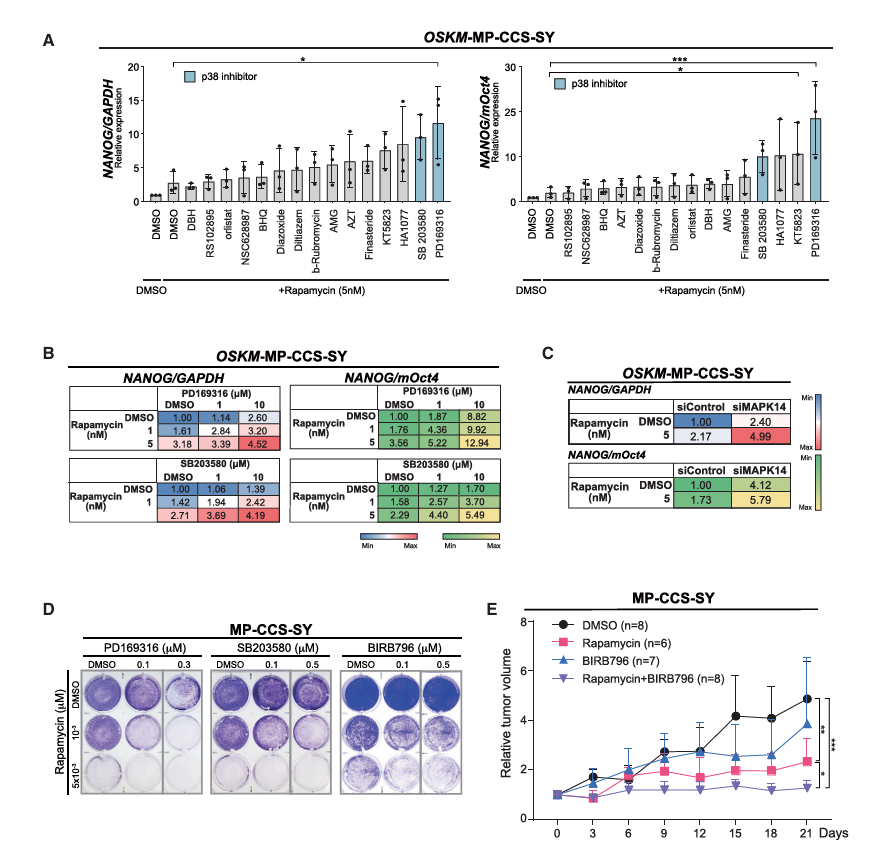
筛选策略: 在rapamycin处理基础上,继续筛选哪种化合物能进一步增强NANOG表达
发现:p38抑制剂(PD169316)+ rapamycin → NANOG表达协同增强
siRNA敲低MAPK14(编码p38α)+ rapamycin → 同样协同效果体内验证(Fig 6E):
MP-CCS-SY异种移植:
DMSO → 肿瘤持续生长
Rapamycin单药 → 中度抑制
BIRB796(p38抑制剂)单药 → 轻度抑制
Rapamycin + BIRB796联合 → 显著抑制(优于任何单药)✓→ 联合用药方案通过重编程抵抗筛选系统被成功鉴定
关键发现
- 癌基因(EWS/ATF1)通过将重编程因子"劫持"到癌症特异性增强子,阻止了癌细胞的重编程——揭示了癌细胞"转录僵硬性"的分子基础
- 这种重编程抵抗是癌基因依赖的 → 抑制驱动癌基因信号可以解除抵抗
- 多种癌症类型(EGFR突变、HER2扩增、BCR-ABL融合、KRAS突变)都存在同样的规律 → 普适性
- 基于NANOG表达的筛选平台比传统增殖/存活筛选更能富集到驱动信号通路抑制剂
- 通过该平台发现mTOR是CCS的驱动通路 → Rapamycin在体内有效抑制CCS生长
- 该平台可用于联合用药筛选 → mTOR + p38抑制剂联合方案协同抑制CCS
- PDC的NANOG响应与患者临床药物敏感性一致 → 精准医疗潜力
证据链
① 癌基因阻止了重编程的早期转录应答(RNA-seq证明)
② 机制:癌基因将重编程因子劫持到癌症增强子(ChIP-seq证明)
③ 抑制驱动癌基因 → 解除重编程抵抗 → NANOG上升(多种癌症验证)
④ 利用NANOG作为筛选指标 → 高效筛出驱动信号通路抑制剂(多种已知靶点验证)
⑤ 应用到CCS → 发现mTOR是EWS/ATF1下游的治疗靶点
⑥ 体内异种移植验证Rapamycin有效 + 联合用药方案
结论:癌细胞对重编程的抵抗性可以被"反过来利用",作为发现癌症治疗靶点的工具与研究室其他工作的关系
这篇论文完成了Project 1的"三部曲"逻辑闭环:
Cell 2014 → 发现问题:表观遗传异常可以独立驱动癌症
↓
Nat Commun 2018 → 理解机制:表观遗传改变与癌基因协同的分子细节
↓
Cell Reports 2022(本篇)→ 解决问题:利用重编程抵抗性发现癌症治疗靶点思路的核心转变:
- 前两篇是"正向"研究:重编程 → 导致癌症(理解疾病)
- 本篇是"反向"应用:癌症 → 抵抗重编程 → 利用抵抗来筛药(治疗疾病)
这种从"发现问题"到"理解机制"再到"解决问题"的完整闭环,正是一个成熟研究方向应有的逻辑链条。面试时如果你能清晰地阐述这条线,会非常有说服力。
面试可能被问到的问题
准备回答
- 这个筛选系统相比传统的药物筛选(看增殖/存活)有什么优势和局限?
- NANOG作为筛选指标有什么局限性?有没有不适用"癌基因成瘾"概念的情况?
- 从CCS中发现的mTOR靶点,临床上是否已有mTOR抑制剂在CCS患者中的试验?
- 这种筛选策略能否应用到更常见的癌症(如结直肠癌、胃癌)?
- 你如何理解"癌细胞身份的僵硬性"(transcriptional stiffness)这个概念?
- 如果你来设计下一步实验,你会怎么改进这个筛选系统?
个人思考
这篇文章更进一步,将重编程的应用前景展示了出来,可以算是一个很有启发性的引用,不过NANOG上升≠真正的重编程,且整个过程都在体外,体内验证仅限于异种移植,这也是未来可以继续完善探索的地方。
从14年到22年的三篇代表性文章可以看出来,研究室对于重编程的重视程度以及专精程度,很厉害👍